METABOLISMO DOS AMINOACIDOS
Destino dos grupos amina
Destino da amonia
Patologia da amonia
Eliminação do carboxilo
Grupos metabolicamente activos
Glicina
Patologia da glicina
Serina
Treonina
Aminoacidos ramificados
Patologia dos aminoacidos ramificados
Lisina
Patologia da lisina
Ãrginina e ornitina
Acido glutamico
Histidina
Patologia da histidina
Prolina e hidroxiprolina
Patologia da prolina e hidroxiprolina
Aminoacidos com enxofre
Patologia dos aminoacidos com enxofre
Fenilalanina e tirosina
Patologia da fenilalanina e tirosina~
Triptofana
Patologia da triptofana
Digestao das proteinas
Metabolismo da hemoglobina
Patologia dos eritrocitos
Sintese dos nucleotidos puricos
Sintese dos nucleotidos pirimidicos
Metabolismo das bases puricas
Patologia das purinas
Formação de desoxiribonucleotidos
Formação de outros nucleotidos
Catabolismo dos acidos nucleicos e nucleotidos
Revisões de conjunto
Capitulo 1
DESTINO DOS GRUPOS
AMINA
Revisões de
conjunto
Nomenclatura
Os aminoácidos são ingeridos na sua quase totalidade como
proteínas, necessitando estas de ser hidrolisadas pelos enzimas digestivos.
1 a 2% das proteínas são recicladas diariamente, dando
origem aos seus aminoácidos constituintes. 75 a 80% destes aminoácidos são
reutilizados para a síntese de novas proteínas, sendo os 20 a 25% restantes
catabolisados.
Os aminoácidos fornecidos em excesso na alimentação não são
aproveitados, sendo catabolisados.
Tendo os aminoácidos uma parte comum (amina e carboxilo) e
uma parte diferente (grupo R) serão considerados separadamente o metabolismo de
cada uma destas partes (metabolismo geral e metabolismo especial)
Desaminação oxidativa
Natureza da reacção
A não ser no caso dos aminoácidos - álcoois ( serina,
treonina) esta desaminação é oxidativa pois a remoção do grupo amina está
associada à perda de dois hidrogénios. A reacção faz-se em duas fases
- Desidrogenação
com formação de uma imina
- Os hidrogénios são captados por uma
flavoproteína.
- Remoção do grupo amina com formação do acido α-cetónico correspondente
D – aminoácido – oxidases
Encontram-se espalhadas por todo o organismo, embora o seu
papel ainda hoje não seja conhecido pois actuam sobre aminoácidos não
existentes no organismo, os da série D.
O seu coenzima é a FAD.

L – aminoácido – oxidases
Embora actuem sobre aminoácidos naturais, a sua concentração nos tecidos é muito
reduzida.
O seu coenzima é a
FMN.
Glicocola–oxidase
É um enzima pouco importante.
Só actua sobre a glicocola, transformando-a em ácido
glioxílico
Glutamico-deidrogenase
É um enzima muito espalhado, de importância metabólica
fundamental.
Desamina o ácido glutâmico em ácido α-cetoglutárico

Utiliza tanto a NAD como a NADP
A reacção é reversivel
Está sujeito a uma regulação alostérica.
O ADP e GDP são
efectores positivos e o ATP e GTP efectores negativos, o que significa que o
enzima é activado quanto o aporte energético é insuficiente e inibido no caso
contrário.
Desaminação
não oxidativa
Nos aminoácidos-alcool ( serina, treonina) formam-se ácidos
cetonicos por desaminação não acompanhada de oxidação porque estes já têm um OH
na sua estrutura

Desamidação
A glutamina e a asparagina têm radicais amida.
A glutaminase e asparaginase catalisam a hidrolise que
liberta este radical

http://ead.univ-angers.fr/~jaspard/Page2/COURS/2N2NH3aaetUree/1N2NH3AAetUree.htm
Transaminação
Conceito
É uma reacção de transferência de um grupo amina de um
aminoacido para um ácido α-cetónico aceitador para originar um outro aminoácido
e um outro ácido α-cetónico

É a reacção mais importante do metabolismo dos aminoácidos.
Pode realizar-se em todos os aminoácidos, excepto a prolina,
lisina e treonina.
Tem como coenzima o fosfato de piridoxal
Nomenclatura
Os enzimas catalisando esta reacção são geralmente
conhecidos como transaminases embora pela nomenclatura da União Internacional
de Bioquímica se devam designar como aminotransferases.
Para designar a aminotransferase, usa-se o nome dos aminoácidos implicados na reacção
Assim o enzima catalisando a reacção:
Ácido glutamico + ácido pirúvico → ácido α-cetoglutárico + alanina
designar-se-ia
como glutamico-alanina aminotransferase,
mas é mais comum usar a designação trivial de transaminases.
Neste case utilizam-se o nome dos compostos do lado esquerdo
da equação
No exemplo anterior o
enzima designar-se-ia transaminase glutamico-pirúvica.
ANIMAÇÕES
Transdesaminação
O contraste entre a fraca actividade das L-aminoacido
desaminases (exceptuando a glutamico-deidrogenase) e a grande actividade das
desaminases são a favor da ideia que estes enzimas possam actuar associados
De facto a maior parte dos aminoácidos é degradada por
desaminação oxidativa indirecta graças à associação
transaminases-glutamico-deidrogenase - é
a transdesaminação


ANIMAÇÕES
Capitulo 2
DESTINO DA AMONIA
No decorrer do metabolismo dos
aminoácidos forma-se o grupo amina que em contacto com a agua se transforma em
amónia
A amónia é tóxica porque desloca
para a esquerda a reacção catalisada pela glutamicodeidrogenase, acarretando
uma menor produção de acido alfa-cetoglutarico com a consequente menor alimentação
do ciclo de Krebs
Vias de eliminação da amónia
- Áminação
de cetoacidos
- Formação
de glutamina
- Formação
de ureia, a via mais importante
Aminação de cetoacidos
Passa-se essencialmente com os
ácidos alfa- cetoglutarico e oxaloacetico

Formação de glutamina
A glutamina comporta-se como uma
reserva de amónia
A reacção é catalisada pela
glutamina sintetase


Síntese da ureia
Bibliografia

Formação do carbamilfosfato
O bicarbonato combina-se com a
amónia e o ATP para formar carbonil-fosfato
A reacção é catalisada pela
carbamil-fosfato sintetase (CPS) I, mitocondrial
Existe uma CPS II, citoplasmica
que entra na síntese das pirimidinas
Formação de citrulina
O carbamilfosfato combina-se com a
ornitina parta dar citrulina pela acção da ornitina-carbamiltransferase
A energia é fornecida pela ligação
fosfato de forte potencial do carbamilfosfato
Formação de arginino-succinato
Forma-se por condensação da
citrulina com o aspartato pela acção da argininosuccinato sintetase
A citrulina atravessa a membrana
mitocondrial pela acção da citrulina-ornitina translocase
Formação de arginina
A arginino-sucinato liase cinde o
arginino-succinato em arginina e fumarato
O fumarato através do ciclo de
Krebs forma o oxaloacetato que por
transaminação regenera o aspartato
Formação de ureia
A argina cinde-se em ornitina, que
entrará num novo ciclo, e em ureia, pela acção da arginase
Locais da síntese
A sintese faz-se parcialmente no
citoplasma e parcialmente nas mitocondrias
Capitulo 3
PATOLOGIA DA AMONIA
Formação patologica da amónia
Causas
Para lá da amónia formada nos tecidos, uma grande quantidade
é formada no intestino pelas bactérias
intestinais.
Esta amónia é absorvida pelo intestino e vai para o fígado
que a remove da circulação pelos mecanismos já estudados, nomeadamente pela
formação de ureia
Quando a função hepática está gravemente alterada ou há um
shunt entre a veia porta e a circulação sistémica a amónia entra na circulação
e produz sinais graves de intoxicação.
Toxicidade da amónia
A amónia é tóxica
pelas seguintes razões..
- Aminação
do acido a- cetoglutarico em
glutamico com a consequente redução
do ciclo de Krebs
- Inibição
da isocitrico deidroghenase com acumulação de acido cítrico que irá inibir
a fosfofrutocinase e portanto a glicolise
- Aumento
da degradação dos coenzimas piridinicos
São assim afectados o ciclo de Krebs, glicolise e cadeia
respiratória
Tratamento
- Diminuição
da ingestão de prótidos
- Diminuição
da actividade bacteriana pela administração de antibióticos
- Estimulação
da ureogenese pela administração de argininna ou carbamilglutamato
- Estes
tratamentos são apenas sintomáticos não actuando sobre a causa da doença
hepática
Erros da ureogenese
Tipos
Os erros da ureogenese são:
- Hiperamoniemia tipo I
- Hiperamoniemia tipo nII
- Citrulinemia
- Aciduria
arginino-sucinica
- Hiperargininemia
A intoxicação pela amónia é mais
intensa quando o bloqueio surge antes da formação de citrulina (hiperamoniemias
I e II ) pois nas outras formas os compostos formados podem formar ligações
covalentes com a ureia
Sintomas
Vómitos, astenia, letargia, atraso
mental
Tratamento
- Alimentação
pobre em proteínas
- Alimentação
fraccionada para evitar grandes subidas de amónia
Capitulo
4
ELIMINAÇÃO DO CARBOXILO
É feita por descarboxilação dos aminoácidos, originando
aminas por descarboxilases especificas, que necessitam de fosfato de piridoxal

As aminas formadas são destruídas por diaminooxidases
Capitulo 5
GRUPOS METABOLICAMENTE ACTIVOS
Transmetilações
Os grupos metilo não são sintetizados no organismo.
São transferidos de moléculas que os contêm (dadores de metilo) para outras que os
aceitam (aceitadores de metilo) por
um processo de transmetilação

Metionina
Precisa de ser activada
na sua forma activa, a S-adenosilmetionina pelo enzima activador da
metionina

A sua desmetilação origina a S-
adenosilhomocisteina
Colina e betaina
Agem como dadores
indirectos na medida em que os metilos libertados vão metilar a
homocisteina

Cria-se assim um ciclo do metilo
O transporte dos metilos
necessita de acido tetrahidrofolico FH4
Transferências de corpos em C1
No decorrer do metabolismo dos
aminoácidos formam-se corpos em C1
- Metilo – CH3
- Metileno
– CH2
- Metenilo - CH
- Formilo O=CH
- Formimino -CH-NH
- Estes
corpos em C1 necessitam de FH4
Tambem se forma CO2, que necessita de fosfato de piridoxal
Formação de FH4
Forma-se a partir do
acido folico pela acção da hidrofolato redutase, na presença de \acido
ascórbico.
Os hidrogénios são fornecidos pelo NADPH
O metotrexato é um inibidor
competitivo da hidrofolico redutase, sendo por essa razão usado como
antitumoral
Os radicais são interconvertiveis
Transporte
Os radicais a ser transportados combinam-se com o N5 ou N10
do FH4
Transamidinação
É a transferência do grupo amidina, catalisada pela
transamidinase
Ciclo do glutamilo
Trata-se da transferência do grupo glutamilo do glutatião ou
glutamina para aminoácidos ou péptidos pela glutamiltransferase
Os gama-glutamil- péptidos estão envolvidos no transporte de
aminoácidos pelo rim
Neste sistema a gamaglutamiltransferase (gGT) associa-se á gGTciclotransterase(gGC)
Regeneração do
glutatião
No rim, a cisteina, glicina e acido glutamico regeneram o
glutatião

GLICINA

Interacções com a
serina
A glicina não é um aminoácido essencial pois pode ser
formada a partir da serina
Por outro lado a sua maior via de degradação é a conversão
em serina
Sistema de clivagem
da glicina
Para a glicina se converter em serina é necessário CHOH que é fornecido pela
clivagem de outra molécula de glicina através do sistema de clivagem da glicina
Desaminação
A glicina oxidase
desamina a glicina em acido glioxilico
Embora a glicina se possa converter em acido glioxilico, a
via normal de degradação do acido glioxilico é
a conversão em glicina
Quando falta o enzima este transforma-se em acido oxalico,
surgindo a hiperoxaluiria primaria, doença grave que causa cálculos renais e
pode levar à morte por insuficiência renal
Síntese do glutatião

Síntese do acido
hipurico
Resulta da combinação da glicina com o acido benzóico
Neoglicogenese
Entra na neoglicogenese pela sua conversão em acido piruvico
Síntese da creatina


Capitulo
7
PATOLOGIA DA GLICINA
Hiperglicinemia
Hiperglicinemia
cetosica
Surge na deficiência em propionil-CoA carboxilase que causa
a acidemia propionica.
Não se sabe porque surge a hiperglicinemia
Como se acompanha de cetose designa-se por hiperglicinemia
cetosica
Hiperglicinemia não
cetosica
Falta o enzima de clivagem da glicina
A forma mais frequente é a neonatal
Surge entre as 6 horas e 8 dias após o nascimento
Há falta de sucção, hipotonia profunda,convulsões
Pode conduzir ao coma e à morte
Nenhum tratamento é conhecido
Nos doentes com formas mais moderadas, os medicamentos que
se opõem à acção da glicina sobre as células neuronais ( estricnina, diazepam,
dextromorfano) podem ter algum efeito
Hiperoxaluria
Causa
Falta a alanina-glioxilato aminotransferase
O enzima encontra-se apenas nos microssomas do fígado
O acido glioxilico não é metabolisado e transfere-se para o
citossol onde é transformado em acido oxalico
Sintomas
O oxalato de cálcio é relativamente insolúvel depositando-de
no rim
Surgem cálculos renais, nefrocalcinose e insuficiência renal
A insuficiência renal agrava-se progressivamente, levando à
morte geralmente antes dos 20 anos
Tratamento
Não há tratamento eficaz
Os transplantes renais não actuam mas descreveram-se bons
resultados com transplantes de fígado e rim
Bibliografia
Capitulo 8
SERINA
Síntese
Origina-se a partir do acido fosfoglicerico, formado na
glicolise

Desaminação
Converte-se em piruvato pela serina deidrase
Descarboxilação
Descarboxila-se em etanolamina, que, por metilações
sucessivas originará a trimetiletanolamina ou colina
Outras actividades metabólicas
- Neoglicogenese
por conversão em acido piruvico
- Síntese
da esfingosina
- sintese
da cisteina
- conversão
em glicina
- síntese
das pirimidinas
Capitulo 9
TREONINA
É um aminoácido essencial
Cisão em glicina e
acetaldeido pela acção da treonina aldolase. O acetaldeido
converte-se em acetilCoA
Desaminação em acido
alfa-cetobutirico que se
descarboxila em acido propionico, originando este o sucinil- CoA
Capitulo 10
AMINOÁCIDOS RAMIFICADOS
(leucina, isoleucina, valina)
São aminoácidos
essenciais.
O catabolismo
destes três aminoácidos segue os mesmos passos, evidentemente com a formação de
intermediários diferentes
Etapes do catabolismo
Transaminação
Formam-se os
ácidos cetónicos correspondentes.
Como a reacção é
reversível, estes cetoácidos podem substituir, na alimentação, os aminoácidos
correspondentes
Descarboxilação
oxidativa dos acidos cetónicos.
Esta reacção é
catalisada por um complexo multienzimático mitocondrial que actua sobre os três
ácidos cetónicos, a desidrogenase dos ácidos cetónicos de cadeia ramificada
(BCKDH) Aleucina origina o isovaleril-CoA,
a isoleucina o metilbutirilCoA e a valina o isobutirilCoA.
A estrutura e sistema de regulação deste enzima é semelhante à
piruvico-deidrogenase.
O enzima é
inactivado quando fosforilado por uma cinase e reactivado por uma fosfatase
cálcio-independente.
Pelo seu lado a
cinase é inibida pelo ADP, ácidos cetónicos de cadeia ramificada, alguns
hipolipemiantes e tioesteres do CoA.
Catabolismo dos
ácidos cetonicos
Após várias
transformações, o isovaleril CoA origina o acetil-CoA e o acido acetoacetico
O metilbutiril CoA
origina o acido propionico que se poderá transformar em sucinil-CoA
O isobutiril-CoA
origina o sucinil-CoA
Acção metabólica
Como consequência
deste catabolismo, a leucina é cetogenica e os outros dois aminoácidos,
glicogenicos
CAPITULO 11
PATOLOGIA DOS AMINOACIDOS RAMIFICADOS
Doença do xarope de acer
Bibliografia
Forma clássica
Falta o complexo
enzimático que descarboxila os ácidos cetonicos, a BKCDH.
Os ácidos
cetonicos ramificados são eliminados pela urina
dando-lhe um cheiro característico, comparado ao do xarope de acer.
A doença é fatal
se não forem removidos rapidamente os ácidos ramificados.
Como a sua
depuração renal é reduzida deve-se recorrer à diálise peritoneal ou à
hemodiálise
Após a recuperação
deve-se instituir uma dieta pobre nestes aminoácidos.
Como estes
aminoácidos são essenciais devem-se adicionar pequenas quantidades à dieta
Quando o aporte de
leucina não atinge o mínimo necessário pode surgir acrodermatite
enteropatica
Forma intermitente
As crianças
aparentemente normais após um stress como febre ou cirurgia desenvolvem a forma
clássica, podendo mesmo ocorrer a morte
O tratamento da
fase aguda é idêntico
Após a
recuperação, embora seja tolerada uma dieta normal, é recomendável uma dieta
pobre nestes aminoácidos
Forma moderada
As manifestações
são insidiosas e ligadas ao sistema nervoso central
O tratamento é o
mesmo
Acidemia
isovalerica
Deficiência da
isovalerilCoA deidrogenase
Nas formas agudas
observa-se vómitos, acidose severa, convulsões e coma
Também existe uma
forma intermitente
O tratamento da
fase aguda incide no tratamento da hidratação, fornecimento de calorias
adequadas de forma oral ou intravenosa, correcção da acidose metabólica pela administração de bicarbonato
Também se deve
remover o acido isovalerico em excesso
Uma solução é a
administração de glicina(250mg/kg/dia) formando-se a isovalerilglicina que tem
uma depuração urinaria elevada
A
carnitina(100mg/kg/dia) também está indicada, por formar
isovalerilcarnitina que é
excretada pela urina
Depois da recuperação está indicada uma dieta pobre em roteínas
e suplementosde glicina e carnitina
Bibliografia
Deficiência em
metilcrotonil-CoA-carboxilase
As manifestações
clínicas são variadas podendo ser fatais com acidose, convulsões e hipotonia
grave
Os episódios
agudos tratam-se com hidratação, infusão intravenosa de glicose e alcalinos.
O tratamento a
longo prazo implica uma dieta pobre em leucina e administração oral de
carnitina
Hipervalinemia
Falta a transaminase da valina
CAPIT ULO XIA
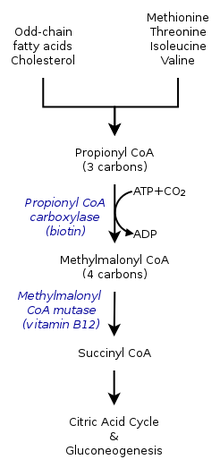
Acidemia propionica
Causas
Defeito da propionato carboxilase cujo coenzima é a biotina
Acumulam-se produtos tóxicos nos gânglios basais
Sintomas
Infarto bilateral dos gânglios basais
Convulsões, vómitos, letargia, desidratação, cetoacidose
Tratamento
Alimentação pobre em proteínas
Carnitina, para melhorar o metabolismo dos ácidos de longa
cadeia
Biotina, coenzima da carboxilase.
Esterilização da flora intestinal, produtora de acido
propionico
Capitulo 12
LISINA
É um aminoácido essencial.
Entra na composição do colagénio após ser convertida em hidroxilisina.
Formação de
ácido glutárico
A lisina transforma-se em ácido glutárico tendo como intermediários os
ácidos α-aminocetoadípico e α –cetoadípico
O ácido glutarico na forma de glutaril-CoA cindir-se-á em malonil-CoA

Formação de sacaropina
É a via catabolica mais
importante
Forma-se por condensação da lisina com o ácido α-cetoglutárico pela acção
lisina- a-cetoglurarico-reductase.
A
sacaropina transforma-se no semialdeido aminoadipico que após vãrias
transformações originará duas moleculas
de acetil-CoA
.
Formação de ácido pipecólico
A lisina através de uma série de
intermediários cicliza-se em acido pipecolico
O ciclo cinde-se para formar o semialdeido a-aminocetoadipico
Este cinde-se em duas moléculas de
acetil-CoA
Capitulo 13
PATOLOGIA DA LISINA
Sacaropinuria
Não se efectua a degradação da
sacaropina por falta da lisina-alfacetoglutarato deidrogenase e da sacaropina
deidrogenase
Lisinemias
Bibliografia
Falta a lisina-cetoglutarato redutase que catalisa a condensação da
lisina com o acido alfacetoglutarico na via da sacaropina
CAPITULO 14
ARGININA
E ORNITINA
Bibliografia

A ornitina por
descarboxilação origina a putrescina e a arginina a agmatina.
Na ureogenese a
arginina cinde-se em ornitina e ureia.
Combina-se com a
glicina para formar a creatinina
CAPITULO 15
PATOLOGIA DA ORNITINA
Ornitinemia
Deficiência em ornitina-aminotransferase
Causa uma entidade clínica bem definida caracterizada por
uma atrofia da coroideia e da retina
Há uma perda progressiva de visão que leva à cegueira na quarta
década da vida
A progressão da doença pode ser travada por restrição em
ornitina e administração de vitamina B6
Capitulo 16
ÁCIDO GLUTAMICO
Bibliografia
Tem uma situação fulcral nas reacções de transaminação, funcionando com
qualquer aminoácido e também em associação com a glutamico deidrogenase nas reacções
de transdesaminação
R1-amino acid + R2-α-ketoacid ⇌ R1-α-ketoacid + R2-amino acid
Alanine + α-ketoglutarate ⇌ pyruvate + glutamateAspartate +
α-ketoglutarate ⇌ oxaloacetate + glutamate
A sua amida, a glutamina, é transportadora de amónia e dadora de NH3
É constituinte do glutatião e dos ácidos fólicos
Participa em certos processos de destoxificação como a combinação com o
ácido fenilacético para originar fenilacetilglutamina, sua via de eliminação
Biosintese
→ Glu + NH3
|
||
→ Glu + Acetate
|
(unknown)
|
|
→ Glu + NADP+ + H2O
|
||
→ Glu + α-keto acid
|
||
→ Glu + NADH
|
||
→ Glu + 5-formimino-FH4
|
||
NAAG
|
→ Glu + NAA
|
Biosintese da prolina


Metabolismo no S.N.C.
No sistema nervoso central o
ácido glutamico é descarboxilado pela c-glutamato
descarboxilase, em ácido c-aminobutírico (GABA) na presença de fosfato de
piridoxal
. O GABA é um neurotransmissor.
O GABA também se pode formar pela
desaminação da putrescina pela diamino-oxidase
GABA e ciclo de Krebs
O GABA pode transaminar-se em
semialdeido succinico pela acção da GABA
transaminase.,indo transaminar o acido alfacetoglutarico para formar acido
glutamico
O semialdeido succinico pode oxidar-se em ácido succinico ou
pela acção da lactico-deidrogenase e dar de novo o GABA
O semialdeido succinico ao converter-se em ácido succcinico,
completa um desvio do ciclo de Krebs pelo qual o ácido α-cetoglutárico em vez
de originar directamente o ácido succinico, fá-lo através do ácido glutâmico,
GABA e semialdeido sucínico


Formação do acido
alfa-glutaramico
- A
glutamina pode transaminar-se em acido alfacetoglutaramico
- Este
acido aparece aumentado mais de 10 vezes nos doentes com coma hepático
- A
sua perfusão no ventrículo lateral de ratos diminui a actividade
locomotora e causa mioclono
Capitulo 17
HISTIDINA
A histidina é um aminoácido essencial, precursor das bases
púricas.
Bibliografia
Descarboxilação
A histidina descarboxila-se em
histamina pela acção da histidina descarboxilase


A histamina
é libertada nos fenómenos alérgicos e estimula fortemente a secreção
gastrica
Desaminação
Desamina-se em acido
imidazol-piruvico
Este pode-se descarboxilar em
imidazol-acetico
O acido
imidazol-acetico tambem se pode produzir pela descarboxilação da histamina
Formação de acido glutamico
É a via mais importante
A histidase desamina não oxidativamente
a histidina em acido urocanico
A urocanase hidrata o acido
urocanico em imidazol-propionico
A imidazol hidrolase cinde o
ciclo imidazol, formando-se o acido formiminoglutamico (FIGLU)
Este cede o formimino ao FH4
formando-se acido glutamico e formimuino-FH4, este ultimo necessário para a
síntese das purinas

Capitulo 18
PATOLOGIA DA HISTIDINA
Histidinemia
É devida
à falta da histidinase
Metade dos casos descritos tem atraso mental e dificuldades
na fala
Na carência de ácido fólico não se forma formimino-FH4,
acumulando-se o FIGLU cujo aumento é considerado como um sinal desta carência.
Doença de Canavan
Na doença de Canavan falta a aspartil acilase, sendo
acompanhada da degenerescência da substancia branca do cerebro
1 em 5000 judeus askhenazi têm esta doença e 1 en 38 são
portadores
Bibliografia
Deficiencia em
urocanase
- Manifesta-se por
dificuldades de aprendizagem e concentração
- Atraso mental
- Atraso de crescimento
- Olhos azuis
Bibliografia
Carnosinemia
Deve-se à deficiência em aminoacilhistidina dipeptidase
Caracteriza-se por:
- Hipotonia
- Atraso mental
- Neuropatia semsorial
- Tremores
- Desmielinização
- Anomalias da matéria
cincenta
- Convulsões mioclonicas
Bibliografia
Capitulo 18
PROLINA
E HIDROXIPROLINA
A hidroxiprolina é um
constituinte importante do colagénio.
Não são aminoácidos essenciais
Têm um papel importante na
estrutura do colagenio
Síntese
Prolina
A prolina sintetiza-se a partir
do semialdeido glutamico que pode ser originado pelo ácido glutamico por
redução ou pela ornitina por transaminação
O semialdeido em seguida
cicliza-se em ácido pirrolidona-carboxilico que depois se reduz em prolina

Hidroxiprolina
Sintese
A hidroxiprolina forma-se por
hidroxilação da prolina, já depois deste aminoácido estar incorporado na cadeia
proteica.
Catabolismo
Prolina
Segue a via inversa à sua síntese
Hidroxiprolina
A maior parte e catabolisada não
na forma livre mas sob a forma de um péptido com hidroxiprolina
Parte da hidroxiprolina é
catabolisada por uma via igual à da prolina
Capitulo 20
PATOLOGIA DA PROLINA E
HIDROXIPROLINA
Bibliografia
Hiperprolinemia
tipo I
Falta a prolina oxidase
Hiperprolinemia tipo
II
Falta a pirrolidona carboxilico deidrogenase
Hidroxiprolinemia
Falta a hidroxiprolina oxidase
Degradação do
colagenio
A hidroxiprolina aumenta nas doenças em que há degradação do
colagenio
Bibliografia
Capitulo 21
AMINOÁCIDOS COM ENXOFRE
Bibliography
Dos vários aminoácidos com enxofre só a
metionina é essencial
A metionina pode-se converter nos outros
aminoácidos com enxofre
A maior parte dos enzimas contem grupos
sulfidrilo da cisteina, que conferem uma função protectora
Interconversões
Embora a metionina seja
essencial, pode formar-se parcialmente por metilação da homocisteina.
A homocisteina e a metionina reagem através
das suas formas activas, a S-adenosilhomocisteina e a S-adenosilmetionina,
respectivamente


A betaina e a colina são fontes importantes
de metilos
Formação da cisteina
A cisteina forma-se por
conjugação da homocisteina com a serina.
Forma-se inicialmente cistationina que
posteriormente se cinde por hidrólise em cisteina e homoserina

A homoserina desamina-se nem
ácido α-cetobutirico que em seguida originará propionil-CoA
Transaminação
A cisteina-glutamato-aminotransferase
transamina a cisteina em ácido sulfinilpiruvico.
Este transforma-se em piruvato e SH2 pela
acção da tiol-pirúvico-transsulfurilase na presença de glutatião reduzido
L-cysteine
+ 2-oxoglutarate = mercaptopyruvate + L-glutamate.
|
||
Metabolismo do SH2
O SH2 é oxidado em SO2 e este em
SO4
O SO4 será excretado ou se
transformará na sua forma activa o fosfoadenosina-fosfosulfito ou PAPS.
A formação de PAPS necessita de ATP
Oxidação
O SH da cisteina pode-se oxidar
em SO3H2 originando o ácido cisteico
O acido cisteico
descarboxilar-se-á em taurina

A cisteina pode oxidar-se em cistina formando-se uma ponte S-S
Se esta oxidação se fizer numa cisteina situada no centro
activo de um enzima, este ficará inactivo
Descarboxilação
A descarboxilação da cisteina
origina a mercaptoetanolamina
Funções do SH na estabilidade
dos enzimas
Muitas enzimas necessitam de um
grupo SH livre para exercerem a sua actividade.
A oxidação do SH inactiva o enzima
Os metais pesados( mercúrio,
arsénio) combinam-se com o SH
inactivando os enzimas que o contêm.
O glutatião reactiva o SH por
redução do S-S.
Nas reacções em que intervém o coenzima A, a
combinação deste com o substracto faz-se no grupo SH que é parte integrante da
estrutura do CoA.
Funções de conjugação
A cisteina pode-se combinar com
halogénios para formar ácidos mercapturicos
A taurina combina-se com os
ácidos biliares para formar p.ex. o ácido taurocólico.
Capitulo 22
PATOLOGIA DOS AMINOACIDOS COM ENXOFRE
Bibliografia
Homocistinemia
Bibliografia
Deficiência em
homocisteina tionase

Acumula-se homocisteina e por remetilação desta metionina
A homocisteina pode reagir com os grupos lisil do colagenio
Sintomas
Observa-se deslocamento do
cristalino após os 3 anos e osteoporose na criança
Nalguns casos o atraso mental é o
primeiro sintoma
Tratamento
Tentou-se como terapêutica a
restrição de metionina e a alimentação
com betaina ou colina
Nalguns casos obtiveram-se bom
resultados com vitamina B6
Actualmente há dados consistentes
sobre a causa ser uma deficiência em folato, sugerindo a administração de
folato como via de tratamento
Defeitos na formação
de metilcobalamina


Cinco defeitos enzimáticos
diferentes podem interferir na formação de metilcobalamina
Os doentes têm vómitos, letargia,
hipotonia e atrasos no desenvolvimento
O laboratório revela uma anemia
megaloblastica
Tratam-se com a administração de
vitamina B12
Bibliografia
Deficiência em metileno- tetrahidrofolato redutase
Na deficiência total há apneia
neonatal e convulsões que podem levar rapidamente à morte
Na deficiência parcial observa-se
um quadro crónico com atraso mental, convulsões,microcefalia e espasticidade
Trata-se com a administração
precoce de betaina
Bibliografia
Homocistinemia e
ateroesclerose
A homocistenemia é um factor de
risco da ateroesclerose
A homocisteina em excesso forma a
sua tiolactona que reage com os aminoacidos livres das LDL provocando a sua
agregação e endocitose pelos macrofagos
Provoca ainda oxidação dos
lipidos e agregação das plaquetas
Hipermetioninemia
Bibliografia
Secundaria
Observa-se em doenças hepáticas
na tirosinemia tipo I e na homocistinuria clássica
Pode observar-se na imaturidade
transitória da metionina adenosiltransferase em recemnascidos alimentados com
dietas ricas em proteínas
Primaria
Deficiencia da adenosina
metiltransferase hepática
Pode haver desmielinização
Cistinuria
Doença autosomica recessiva da
reabsorção tubular dos aminoacidos
dibasicos cistina, ornitina, lisina e arginina
Foi descrito uma alteração
semelhante na mucosa intestinal mas não tem consequência porque estes
aminoácidos são sintetizados no organismo
A cistina é relativamente
insolúvel, podendo nos homozigotos,
precipitar e formar cálculos renais
Bibliografia
Cistinose
É uma doença muito rara mas de grande gravidade
Há deposito de cistina em
muitos órgãos e tecidos
A lesão tubular renal produzida pela cistina pode causar a
doença de Fanconi
A morte é precoce
Na fig. 22.1 resumimos as causas metabólicas destes erros do
metabolismo
Fig 22.1
Causas metabólicas
Bibliografia
CAPITULO 23
FENILALANINA E TIROSINA
Bibliografia
Transformação da fenilalanina em tirosina
É a via normal do metabolismo da fenilalanina.
Esta transformação faz-se pela acção da fenilalanina-hidrolase.
A fenillalanina hidrolase tem como coenzima a biopterina,
que é uma biopteridina

As biopterinas têm grande semelhança estrutural com as
flavinas e podem participar nas oxidações biológicas.
A forma activa da biopterina é a 5,6,7,8
tetrahidropterina que se forma a partir da 7,8-dihidropterina. Esta
transformação é catalizada pela dihidropterina redutase, NADH dependente
Este conjunto de reacções é conhecido como NIH shift por
ter sido caracterizado por cientistas do NIH
Catabolismo da tirosina
Transformação em
ácido p-hidroxifenilpiruvico pela
acção da tirosina transaminase.
Formação de ácido homogentísico
pela acção da p-hidroxifenilpiruvato dioxigenase, sendo o oxigénio
fornecido pelo oxigénio molecular.
O enzima tem um Fe++
que forma um complexo com o oxigénio molecular.
A homogentisico-1,2-dioxigenase
catalisa a formação do ácido maleilacetico.
A maleilacetoacetato-isomerase
catalisa a isomerização em ácido fumarilacetico
Em seguida a fumarilacetoacetase origina
acetoacetato e fumarato


Síntese de outras substancias
A tirosina é necessária para a síntese das hormonas
tiroideias, da alanina e nor-adrenalina e das melaninas

Capitulo 24
PATOLOGIA DA
FENILALANINA E TIROSINA
Alcaptonuria
Bibliografia
O acido homogentisico acumula-se nos tecidos e no sangue e
depois passa para a urina, dando-lhe uma cor escura

A oxidação e polimerização deste acido produz um pigmento, a alcaptona
A alcaptona deposita-se nas cartilagens dando-lhes uma cor
ocre, donde o nome ocronose


A esclerótica tem uma cor
azulada


Pode acompanhar-se de artrite


Não há tratamento efectivo
Tirosinemia
Bibliografia
Tirosinemia tipo I
Bibliografia
Descrição
É a deficiência em fumaril-acetato hidrólase

A acumulação de maleilacetato e
fumarilacetato que são agentes alquilantes, provocam alquilação de DNA
Observa-se insuficiência hepatica
e renal, raquitismo e polineuropatia
Tratamento
Nalguns doentes obtêm-se bons
resultados com dietas pobres em tirosina e fenilalanina
Na maior parte dos casos trata-se
com um inibidor da hidroxifenilpiruvato dioxigenase
Tirosinemia tipo II
Bibliografia
Não se faz a transaminação da
tirosina em ácido p-hidroxifenilpirúvico devido à deficiência em tirosina
aminotransferase
Lesões nos olhos e pele



Atraso mental
Responde bem a dietas pobres em
tirosina e fenilalanina
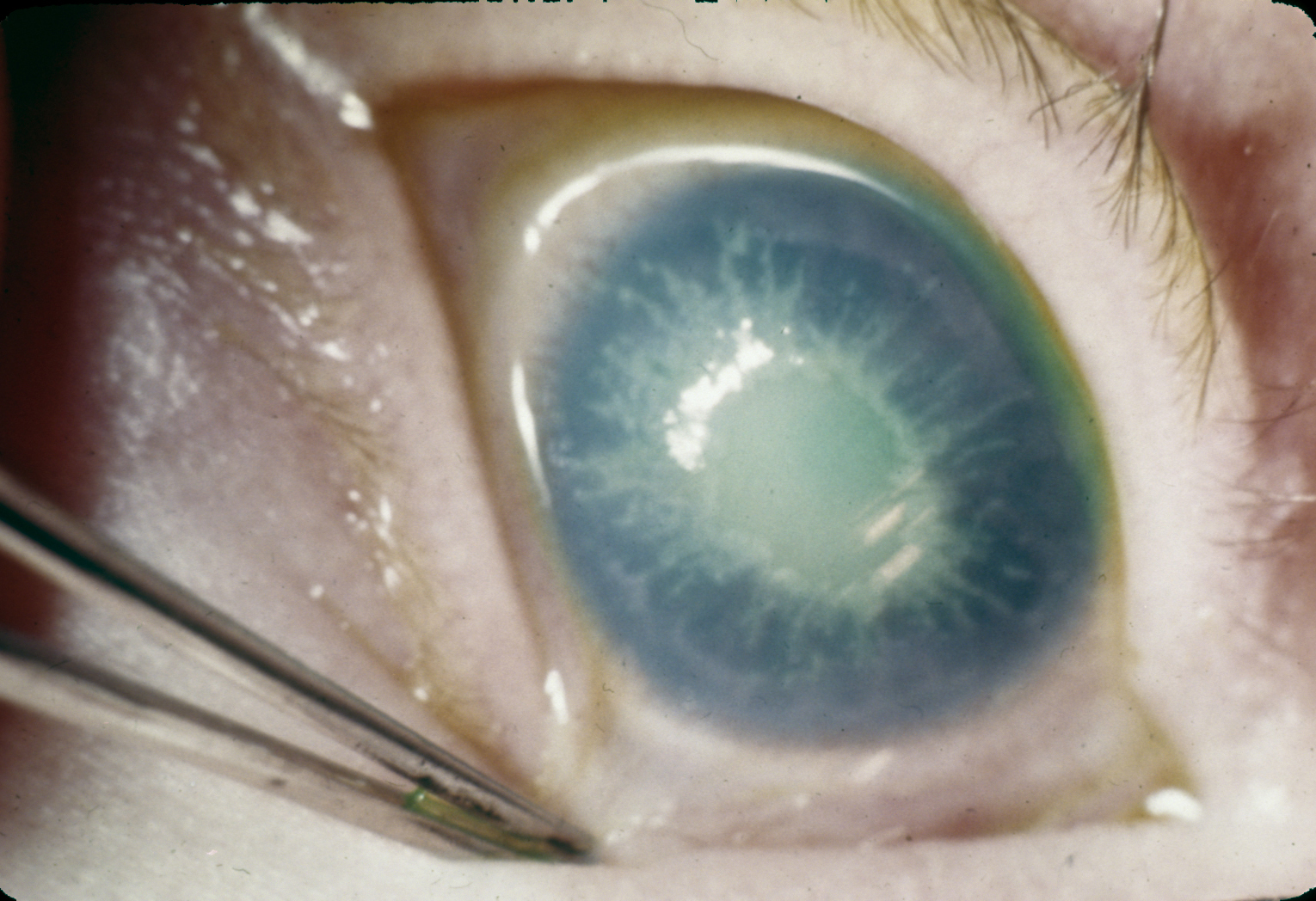
Albinismo
Bibliografia
Falta a tirosinase nos melanocitos, não se formando melanina
A pigmentação da pele, cabelos e
íris é reduzida


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A fraca pigmentação da íris causa
fotosensibilidade
A fraca pigmentação da pele está
associada com uma maior incidência do cancro da pele
A tirosinase envolvida na síntese
das catecolaminas é um isoenzima diferente e não é afectada pelo gene do albinismo
Oligofrenia fenilpiruvica
É o erro mais frequente
Bibliografia
Falta de tirosina-oxidase
A fenilalanina não se transforma
em tirosina, em geral por falta da tirosina-oxidase.
A fenilalanina converter-se-á
alternativamente em ácido fenilpiruvico pela acção de uma transaminase.

Falta de formação de tetrahidropteridina
Conceito
1 a 2% dos doentes com hiperfenilalaninemia
não sintetizam a tetrahidropteridina(BH4)
Estes doentes têm os mesmos
sinais e por isso muitas vezes são diagnosticados como deficientes em
tiroxina-oxidase
Todavia nestes doentes os
sintomas mantêm-se após a supressão da fenilalanina na alimentação
Acção da BH4
A BH4 é também coenzima da triptofana-hidroxilase, necessária para a
síntese da dopamina e da serotoni e também do enzima que catalisa a produção do
acido nítrico a partir da arginina.
Defeitos enzimáticos
A BH4 é sintetisada a partir da
GTP
Descreveram-se quatro
deficiências enzimáticas nesta via
Rastreio
A fenilcetonuria afecta 1 em cada
10.000 crianças.
No nascimento não surgem sinais
mas eles surgem precocemente caminhando rapidamente para uma oligofrenia grave.
Por esta razão deve-se fazer o
seu rastreio em todos os recemnascidos
Fenilcetonuria nas
grávidas
As gravidas com
hiperfenilalaninemia correm um risco grave de ter filhos com oligofrenia
fenilpiruvica
Devem fazer dieta para manter a
fenilalanina abaixo de 6mg%
Tratamento
Correcção da hiperfenilalaninemia
Devem-se administrar alimentos
pobres ou isentos em fenilalanina
A dieta deve ser iniciada
imediatamente após o diagnostico
A dieta é obrigatória para
valores acima de 6mg%
Como a fenilalanina é um aminoácido
essencial, a dieta deve ser monitorizadas para os valores se manterem
dentro do normal (2-6 mg%)
Administração de BH4
A administração de BH4 em doentes
com defiociencia da tiroxina-oxidase pode ser útil
Nos doentes com deficiência em
BH4 a administração de BH4 é obrigatória
Os deficientes em GTP
ciclohidrolase respondem com doses
menores (5-10 mg/kg/dia) que os deficientes em dihidropteridina redutase\(20
mg/kg/dia)
Administração de neurotransmissores e acido folico
Nos deficientes em BH4 recomenda-se
a administração de dopamina e serotonina mesmo que o BH4 normaliza os teores em
serotonina pois que o BH4 não penetra facilmente o tecido cerebral
A suplementação em acido folico
está recomendada na deficiência em dihidropteridina redutase
Deficiência da BH4 com fenilalanina normal
É muito rara
Deve-se à deficiência em GTP
ciclohidrolase
Manifesta-se por distonias
musculares e sinais de Parkinson
Trata-se com DOPA associada ao
inibidor proteico da dopa-descarboxilase
Capitulo 25
TRIPTOFANA
É um aminoácido essencial.
Participa na síntese de compostos
de interesse biológico como a serotonina e a nicotinamida
Bibliografia
Síntese da serotonina , nicotinamida e
melatonina

{kind=link}
Serotonina
A serotonina é a
5-hidroxitriptamina
A triptofana é hidroxilada em
hidroxitriptofana pela triptofana
hidroxilase
A DOPA descarboxilase
descarboxila a hidroxitriptofana em serotonina
É eliminada pela urina como acido
5-hidroxiindolacetico
A serotonina é sintetisada nas células cromafins
Nicotinamida
As primeiras etapas do
catabolismo da triptofana são comuns à síntese da nicotinamida
A triptofana pirrolase cinde na
presença de oxigénio o núcleo da
triptofana
formando formilquinurenina
Esta ao perder a alanina origina
a quinurenina pela acção da quinurenina formilase
A quinurenina pode transaminar-se
e ciclizar-se em ácido quinurénico ou oxigenar-se em hidroxiquinurenina que por
desaminação origina o ácido xanturénico.
Muitos das enzimas intervindo
nestas reacções necessitam de vitamina B6 e por esta razão na deficiência nesta vitamina, aparecem
aumentados na urina alguns metabólitos.
Nomeadamente o ácido xanturénico, a
quinurenina e a hidroxiquinurenina, têm aumento particularmente notável após
uma sobrecarga em triptofana.
Em seguida a hidroxiquinurenina
pela acção da quinureninase perde a alanina para dar ácido
β3-hidroxiantranalinico que se transformará em nicotinamina
A hidroxiquinurenina não
metabolisada em nicotinamida transforma-se em ácido picolinico que, após uma série
de transformações originará o acetoacetilCoA

Catabolismo pelas
bactérias intestinais
No intestino as bactérias
intestinais removem o acido piruvico formando indol
O indol é eliminado pela urina
conjugado com o acido sulfúrico ou o acido glicuronico
Nas fezes é eliminado o seu
derivado natural, o escatol
CAPITULO 26
PATOLOGIA DA
TRIPTOFANA
Bibliografia
Doença de Hartnup
Bibliografia
Absorção reduzida e excreção aumentada de triptofana, devido
a um defeito de transporte dos aminoácidos neutros
Como consequência, a síntese de nicotinamida diminui
A manifestação clínica mais importante é a fotosensibilidade
cutanea
Se o aporte alimentar de nicotinamida for reduzido haverá
sinais de pelagra
Trata-se com a administração de nicotinamida ou acido
nicotínico e uma alimentação rica em proteínas
Esquizofrenia
Alguns autores
propuseram considerar a esquizofrenia como uma doença do metabolismo da
triptofana e por esta razão aconselham
para o seu tratamento dietas pobres em triptofana
Pelagra
A vitamina P.P . é
a única vitamina que é sintetizada no organismo , mas a síntese tem que ser
complementada pela alimentação. Quando a via sintética é inibida por certos
tóxicos como as aflatoxinas surgr a
pelagra
Outras doenças
Descreveram-se
acumulação de plaquetas em depressões e
no cérebro na porfiria intermitentev aguda
Capitulo 29
DIGESTÃO DAS PROTEINAS
Classificação dos enzimas proteoliticos
Enzimas
intracelulares
- Encontram-se
nos lisossomas
- Não
intervêm na digestão
- São
responsáveis pelos fenómenos de autolise post-mortem
Enzimas
extracelulares
- Encontram-se
nos sucos digestivos
- Dividem-se
em exo e endopeptídases

{kind=link}
Endopeptidases
- Hidrolisam
ligações péptido situadas no interior da cadeia
- Existem
como pró-enzimas
Pepsina´
Bibliografia
http://www.sigmaaldrich.com/life-science/metabolomics/enzyme-explorer/analytical-enzymes/pepsin.html
Activação
·
Existe sob uma forma inactiva, o pepsinogenio
·
Em meio acido o pepsinogenio cinde-se num
péptido com cinco aminoácidos e o complexo pepsina-inibidor
·
Em meio mais acido, a pepsina liberta-se do
inibidor
·
Em meio alcalino esta reacção iinverte-se
·
A pepsina formada também hidrolisa o
pepsinogenio – autocatalise

http://www.sigmaaldrich.com/life-science/metabolomics/enzyme-explorer/analytical-enzymes/pepsin.html

Ponto de acção
- Actua
sobre ligações péptido em que a amina pertence a um grupo aromático(
fenilalanina ou tirosina)
- A
uma velocidade menor cinde também ligações leucina-valina e leucina-acido
glutamico
{kind=link}
Tripsina
Bibliografia
- Resulta
da activação do tripsinogenio, sintetizado no pâncreas
- A
activação do tripsinogenio é processada pela enterocinase na presença de
cálcio,libertando-se tripsina e um hexapeptido
- A
tripsina formada activa o tripsinogenio por autocatalise

- Actua
sobre ligações péptido em que a
arginina e a lisina participam pelos seus carboxilos
- O
soro contem um inactivador da tripsina
Quimotripsina
Bibliografia
·
É activada pelo tripsinogenio


{kind=link}
·
A sua especificidade é menor
·
Hidroliza ligações péptido em que estejam
implicadas pelo carboxilo a tirosina, fenilalanina, triptofana, metionina ou
leucina.
·
Hidroliza ligações péptido em que estejam
implicadas pelo carboxilo a tirosina, fenilalanina, triptofana, metionina ou
leucina.

{kind=link}
Exopeptidases
- Hidrolisam
ligações péptido situadas no topo da cadeia
Carboxipeptidases
·
Atacam a ligação péptido mais próxima do
carboxilo terminal
·
A carboxipeptidase A actua sobre aminoácidos
aromáticos, sendo a actividade máxima com a fenilalanina
·
A carboxipeptidase B actua sobre aminoácidos
básicos
·
Não actuam sobre a prolina ou a hidroxiprolina
·
Existem como pró-enzimas, activados pela tripsina
Aminopeptidases
- Actuam sobre a extremidade próxima da amina terminal
Dipeptidases
- Actuam
sobre dipeptidos
- Necessitam
de iões metálicos bivalentes
- Conhecem-se:
Glicil-glicina
dipeptidase
Prolinase
- Só actua se a amina terminal pertencer à prolina
Prolidase
- Só actua se o carboxilo terminal pertencer à prolin
Absorção
- As
proteínas e os péptidos provenientes de hidrolises parciais são degradados
em aminoácidos pelas proteases pancreáticas e pelos enzimas das células intestinais e em seguida
absorvidos por transporte activo

Patologia da digestão dos protidos
Redução das áreas
absortivas
Doença celíaca
- Sensibilidade
à gliadina existente no glúten
- Causa
uma atrofia dos vilos, em especial no intestino delgado proximal, com a
consequente deficiência de absorção
- Em
geral desaparece com a supressão do glúten na alimentação
- Aparece
usualmente no primeiro ano de vida com dificuldades no crescimento e diarreia
Sprue
- A atrofia
dos vilos não responde ao glúten
- Poderá
ter uma causa bacteriana porque às vezes esta situação responde a
antibióticos de largo espectro
- O
folato parece ser útil
Cirurgia
- Grandes
ressecções do intestino delgado reduzem grandemente a área absortiva
Inflamação
- A
inflamação do intestino como na doença de Crohn dificultam a absorção intestinal
Gastrectomia
- Não
se efectua no estômago a mistura dos alimentos com pepsina e acido
clorídrico
- Haverá
uma actividade enzimática diminuída no intestino
- Há
também um aumento de transito no intestino
Pancreas
Fibrose quistica do
pâncreas
- É
uma doença autossomica recessiva do transporte dos cloretos, afectando as
secreções das glândulas exocrinas
- Não
são sintetizados os enzimas pancreáticos
- Digestão
proteica deficiente, levando a sindromas de má absorção
Ablacção da cabeça do pâncreas
- 50%
das proteinas ingeridas são eliminadas pelas fezes
Pancreatite aguda
hemorrágica
- Necrose
das células pancreáticas associada com a <libertação de enzimas para o
espaço retroperitoneal e corrente sanguínea
- A
existência de suco pancreático na cavidade abdominal provoca dor abdominal
intensa e shock
- A
tripsina libertada e activada pela necrose do pâncreas provoca a
autodigestão da cabeça do pâncreas
- Cria-se
assim um ciclo vicioso:quanto mais é destruído o pâncreas, mais enzimas se
libertam
Pancreatite crónica
- É
mais frequente em alcoólicos
- Pode
haver intolerância à glicose pela redução do numero de ilhéus de
Langerhans
- Há
uma diminuição da actividade dos enzimas pancreáticos
Capitulo 30
METABOLISMO DA HEMOGLOBINA
Estrutura
A hemoglobina é um pigmento tetrapirrolico
O pirrol é um
heterociclo pentagonal com azoto

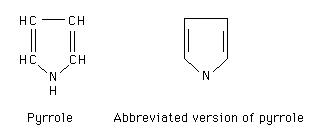
A porfina é uma molécula teórica concebida como a reunião de quatro
pirrois

A introdução de
alguns grupos substituintes nas porfinas forma as porfirinas
Porfirinas
Na hemoglobina existe a protoporfirina IX


A combinação desta com o ferro forma o heme
{kind=link}

A
proteína da hemoglobina é a globina.
É
composta por quatro subunidades contendo hemes
– duas cadeias a
e duas cadeias b

A
sequência de aminoácidos de cada cadeia é determinada geneticamente, levando
qualquer mutação do gene a uma anomalia da estrutura.
Em
cada subunidade, tal como na mioglobina, existe uma bolsa para receber o heme.
Este
une-se pelo seu átomo de ferro que por ter seis valências, dispõe de duas
valências livres para lá das que utilizou para se combinar com os pirrois.
Na cadeia b uma destas valências liga-se à
histidina 92, proximal, e a outra à histidina 63 distal através de uma molécula
de água .
Na
cadeia alfa, as histidinas
envolvidas são as 87 e 58.


Diversidade
estrutural
No
decorrer da vida humana formam-se diferentes formas de hemoglobina.
Na
vida adulta a forma quase exclusiva é a hemoglobina a2b2.
Todavia
desde a concepção até aos primeiros tipos de vida aparecem e desaparecem várias
formas de hemoglobina.
A
hemoglobina fetal contém duas cadeias a e duas cadeias que irão desaparecer,
as cadeias d – é
portanto a2d2.
No
embrião encontram-se as cadeias e e z.
No
adulto 2% da hemoglobiina é constituída
pela hemoglobina A2 de constituição a2d2(

cortesia do prof Christian
Binet
Síntese
O heme é sintetizado por uma série de reacções, umas nas
mitocôndrias, outras no citossol


Cortesia de Joyce Diwan

A primeira reacção é a síntese do
acido deltaaminolevulínico pela reacção do succinil-CoA com a glicina,
catalisada pela deltaminolevulínico
sintetase ou ALA-sintetase
A ALA sintetase encontra-se nas
mitocôndrias, enquanto que o sucinil CÔA é sintetizado no citoplasma,
devendo portanto ser transportado para
as mitocôndrias
O ALA é transportado para o citoplasma para se condensar com outro
ALA para dar o porfobilinogénio
O porfobilinogénio irá sofrer uma
série de transformações para originar a protoporfirina IX
A ferroquelatase irá introduzir o
ferro na protoporfirina IX para originar o heme
Regulação
O enzima chave é a ALA sintetase
O seu principal inibidor
alostérico é o heme
Os esteroides e alguns
medicamentos podem estimular este enzima
Transporte do
oxigénio
Quando o sangue pobre em oxigénio passa pelos pulmões, este
recebe o oxigénio que se difundiu pelos pulmões
Nos eritrocitos o oxigénio, oxigena mas não oxida a
hemoglobina ( o ferro continua bivalente) devido a uma alteração da conformação
da hemoglobina, que se transforma em oxi-hemoglobina
Nos capilares dos tecidos
passa-se o processo inverso – a oxihemoglobina cede o oxigénio aos
tecidos e transforma-se em desoxihemoglobina
Estes fenómenos passam-se porque
a desoxihemoglobina tem maior afinidade para o O2 ( efeito Haldane) e H+
(efeito Bohr)

44
 |
|
|
|
Figure 7
This is a schematic diagram of the
flow of blood through the circulatory system, showing the sites of O2/CO2
exchange in the body.
Note: The components of this diagram are not drawn
to scale.
|
|
cortesia de R.Frey

{kind=link}
Transporte do
anidrido carbónico
O CO2 entra e sai da célula difundindo-se através de canais
transmembranarios
Dentro do glóbulo metade combina-.se com a hemoglobina dando
carbohemoglobina.
A outra metade converte-se em bicarbonato e H+ pela acção da
anidrase carbónica
O bicarbonato difunde-se para o plasma
O H+ combina-se com a hemoglobina que funciona como tampão

Cortesia de Chemical Society
Ciclo de
Rapaport-Luebring ou ciclo do fosfoglicerato
No eritrocito encontra-se o ácido 2,3– bisfosfoglicérico
As suas cargas negativas unem-se às cadeias de carga
positiva da hemoglobina facilitando a expulsão de oxigénio para os tecidos
Forma-se a partir do acido 1,3-bisfosfoglicérico pelo ciclo
de Rapaport-Luebring
Aumenta em populações
vivendo em altas altitudes devido à falta de oxigénio, em situações de anoxia,e
em doenças crónicas em que haja má distribuição de oxigénio e em anemias graves

BIBLIOGRAFIA
Ilustrações-
hemoglobina
Catabolismo do heme
Ciclo de Rapoport-
Luebering
Heme
Ilustrações –
estrutura do heme
Porfirinas
Síntese do heme
Capítulo 31
PATOLOGIA DOS ERITROCITOS
Anemia
É a redução da capacidade do sangue em transportar oxigénio,
devendo-se a diminuição dos eritrocitos, baixo teor em hemoglobina e
hemoglobinas anormais
Diminuição dos
eritrocitos
Anemias hemorrágicas, por perda de sangue
Anemias hemolíticas por destruição precoce dos eritrocitos
Anemias aplasticas por diminuição da eritropoiese
Baixo teor em
hemoglobina
Anemia por perda de ferro ou microcitica. Chama-se
microcitica por os glóbulos serem pequenos e pálidos
Anemia dos atletas
Em períodos de treino intensivo, o volume sanguíneo pode
aumentar até 15%, diminuindo a quantidade de eritrocitos por unidade de volume
Anemia perniciosa por falta de vitamina B12
Hemoglobinas anormais
Conceito
Desde que em 1949 PAULING e ITANO descreveram
que a drepanocitose era devida à existência de uma hemoglobina anormal , a
hemoglobina S, nasceu pela primeira vez a noção que a doença poderia ser devida
à alteração da molécula
Abriu-se assim um novo domínio de estudo, a
patologia molecular
A partir daí descreveram-se inúmeras
hemoglobinas anormais.
Classificação
Conforme a natureza da
anormalidade podemos classificà-las em:
- Alterações
da estrutura primária
- Diferente
combinação das cadeias
- Diferente
repartição das variedades de hemoglobina
Alterações da estrutura primária
As
mais frequentes são a hemoglobina S e as hemoglobinas M
Hemoglobina
S
Na
hemoglobina S houve uma substituição em b6 do ácido glutâmico
pela valina.
Esta
nova valina fica muito próxima da valina em 1 o que leva estas duas valinas a
aproximarem-se pelas forças de van der Waals e a ciclizar a extremidade da
cadeia, ciclização estabilisada por uma ligação ponte de hidrogénio entre o
azoto da treonina 4 e o carboxilo da valina 1.
Esta
ciclização explicaria a formação de glóbulos em foice (falciformação) e a sua
agregação com outras moléculas de globina
Hemoglobinas
M
.Parte
da hemoglobina oxida-se em meta-hemoglobina(ferro trivalente) que não
transporta oxigénio mas em condições normais, a meta-hemoglonina redutase reduz
de novo a meta-hemoglobina
As
hemoglobinas M manifestam-se pela formação de
metahemoglobinas anormais e que persistem porque não se reduzem em hemoglobina
pela acção dos sistemas redutores existentes no organismo o que as impede de
ceder oxigénio aos tecidos.
Devem-se
à mutação de um aminoácido numa zona vizinha à bolsa onde se anicha o heme.
As
mais comuns resultam da substituição de uma histidina(a58, a87 ou b63) pela
tirosina ou da b- valina 67 pelo acido glutâmico.
Diferente
combinação de cadeias
Ausência de cadeias
Nalgumas
destas hemoglobinas há a ausência de
cadeias A.
Nesta situação o seu lugar é ocupado por outra
hemoglobina, no adulto pela b( hemoglobina
b4 ou hemoglobina H)e no feto
pela g (hemoglobina g4 ou hemoglobina de Bart).
Cadeia mista
Nas
hemoglobinas de cadeia mista há um crossing-over entre genes
responsáveis pela síntese de cadeias diferentes.
Na hemoglobina Lepore há duas cadeias a
ligadas a duas cadeias anormais formadas
pela reunião da metade N-terminal da d com a metade C-terminal da b.
Porfirias
Deficiência de um dos enzimas da síntese das porfirinas.
Como levam a uma síntese diminuída do heme deixa de haver a inibição alosterica da
ALA-sintetase
Resulta a acumulação de precursores das porfirinas (ALA,
PBG) e porfirinas
Quando há aumento de precursores, as manifestações são
neurológicas
Quando as porfirinas se acumulam o principal sinal é a
fotosensibilidade – as porfirinas absorvem luz e ficam excitadas, induzindo a
formação de radicais livres.
Hemoglobina
glicosilada
A
hemoglobina A1 reage espontaneamente com a glicose para formar a hemoglobina
glicosilada ou hemoglobina A1.
Normalmente
a concentração desta hemoglobina é muito baixa mas na diabetes pode atingir 12%
da hemoglobina total ou mais.
Hemoglobina
S
Na
hemoglobina S houve uma substituição em b6 do ácido glutâmico
pela valina.
Esta
nova valina fica muito próxima da valina em 1 o que leva estas duas valinas a
aproximarem-se pelas forças de van der Waals e a ciclizar a extremidade da
cadeia, ciclização estabilisada por uma ligação ponte de hidrogénio entre o
azoto da treonina 4 e o carboxilo da valina 1.
Esta
ciclização explicaria a formação de glóbulos em foice (falciformação) e a sua
agregação com outras moléculas de globina
Hemoglobinas
M
.Parte
da hemoglobina oxida-se em meta-hemoglobina(ferro trivalente) que não
transporta oxigénio mas em condições normais, a meta-hemoglonina redutase reduz
de novo a meta-hemoglobina
As
hemoglobinas M manifestam-se pela formação de
metahemoglobinas anormais e que persistem porque não se reduzem em hemoglobina
pela acção dos sistemas redutores existentes no organismo o que as impede de
ceder oxigénio aos tecidos.
Devem-se
à mutação de um aminoácido numa zona vizinha à bolsa onde se anicha o heme.
As
mais comuns resultam da substituição de uma histidina(a58, a87 ou b63) pela
tirosina ou da b- valina 67 pelo acido glutâmico.
Diferente
combinação de cadeias
Ausência de cadeias
Nalgumas
destas hemoglobinas há a ausência de
cadeias A.
Nesta situação o seu lugar é ocupado por outra
hemoglobina, no adulto pela b( hemoglobina
b4 ou hemoglobina H)e no feto
pela g (hemoglobina g4 ou hemoglobina de Bart).
Cadeia mista
Nas
hemoglobinas de cadeia mista há um crossing-over entre genes
responsáveis pela síntese de cadeias diferentes.
Na hemoglobina Lepore há duas cadeias a
ligadas a duas cadeias anormais formadas
pela reunião da metade N-terminal da d com a metade C-terminal da b.
Porfirias
Deficiência de um dos enzimas da síntese das porfirinas.
Como levam a uma síntese diminuída do heme deixa de haver a inibição alosterica da
ALA-sintetase
Resulta a acumulação de precursores das porfirinas (ALA,
PBG) e porfirinas
Quando há aumento de precursores, as manifestações são
neurológicas
Quando as porfirinas se acumulam o principal sinal é a
fotosensibilidade – as porfirinas absorvem luz e ficam excitadas, induzindo a
formação de radicais livres.
Hemoglobina
glicosilada
A
hemoglobina A1 reage espontaneamente com a glicose para formar a hemoglobina
glicosilada ou hemoglobina A1.
Normalmente
a concentração desta hemoglobina é muito baixa mas na diabetes pode atingir 12%
da hemoglobina total ou mais.
Como o tempo médio de vida dos glóbulos vermelhos é de 120 dias,
a hemoglobina glicosilada é um bom índice dos teores em glicose dos últimos
dois meses.ida
Poliglobulia ou
policitemia
Há
aumento do número de eritrocitos
Aumenta
a viscosidade do sangue e diminui a velocidade da circulação
A policitemia primária ou doença de Vaquez deve-se a um
cancro da medula
As
policitemias secundarias podem
resultar de numa adaptação à falta de oxigénio ou à produção excessiva de
eritropoietina
BIBLIOGRAFIA
Destruição dos eritrocitos
Os eritrocitos têm uma vida útil de 120 dias, sendo
destruídos nos pequenos vasos e no baço
Os macrofagos fagocitam os eritrocitos, fragmentam-nos, o
heme separa-se da globina e o ferro é recuperado
O restante do heme é degradado em bilirrubina
A bilirubina é degradada em estercobilina,em urobilina,
incolor, eliminada pela urina e estercobilina, corada, que dá cor às fezes
HEMOGLOBINA
Globina
Ferro---- Transferrina
Biliverdina
H+ NADPH + H+
Biliverdina redutase
Bilirubina
Acido glicuronico
Bilirubina conjugada


{kind=link}
Capitulo 32
SINTESE DOS
NUCLEOTIDOS COM PURINAS
Precursores do núcleo purico
As purinas formam-se a partir dos seguintes precursores
- Glicocola
– C4,C5,C7
- Formil-FH4
– C2,C3
- Glutamina
– N3,N9
- Acido
aspartico – N1
- CO2
– C1
Síntese do IMP
É o primeiro nucleotido a ser
formado
Os outros formam-se a partir dele
Formação do fosforibosilpirofosfato(PRPP)
È a forma activa da ribose
A PRPP sintetase catalisa a combinação da fosforibose com o ATP
Formação do ribotido de glicinamida
A glutamina
cede a amida ao PRPP para formar a fosforibisilamina. O enzima é a PRPP
amidotransferase
A fosforibosilamina reage com a
glicina na presença de ATP para dar o
ribotido de glicinamida O enzima é a fosforibosilglicinamida sintetase
Transferencia de um carbono
O formil-FH4
transfere um carbono, formando-se o ribotido de formilglicinamida O enzima é a
formilglicinamida formiltransferase
Amidinação
A glutamina
cede uma amida para formar o ribotido de formilglicinamidina
É necessário
ATP
Formilação em ribotido de formilglicinamida
A formilação é
feita pelo formil FH4
Ciclização
O ciclo fecha-se formando-ser o ribotido de 5-aminoimidazol
O enzima é a
5-imidazol sintetase
E necessário
ATP
Visão global destas etapas
Ribose
PRP NH2
Glutamina
Fosforibosilamina
Glicina
Ribotido da
glicinamida
FH4
Ribotido da
formilglicinamida
NH2
Glutamina
x
Ribotido da formilglinamidina
Encerramento do ciclo
Ribotido do
amino-imidazol
Carboxilação
O 5-aminoimidazol é carboxilado no acido 5-aminoimidazolcarboxilico
Amidação
O ribotido
amida-se por combinação com o acido aspartico para se formar uma
carboximida, o ribotido da
5-amino-4-imidazol-sucino-carboximida
Perda do acido fumarico
A carboxiamida
perde o acido fumarico para dar o ribotido da 5- aminoimidazolcarboxiamida
Formação do IMP
Forma-se o
5-formamidoimidazol-4-carboxiamida-ribonucleotodo pela reacção com o formil-FH4
Este
desidrata-se em seguida formando o IMP
Síntese do AMP
Formação de acido adenilosuccinico
O IMP
combina-se com o acido aspartico para dar o acido adenilosuccinico
O enzima e a
adenilosuccinatosintetase
A energia para a síntese é
fornecida pela hidrolise do GTP
Formação do AMP
- A
adenilosuccinato é hidrolisado em acido fumarico e AMP
- O
enzima é a adenilosuccinatoliase
Formação do XMP
O IMP é hidrolisado em XMP pela IMP deidrogenase
Formação do GMP
Forma-se pela
transferência de uma amida da glutamina para o
XMP
O enzima é a XMP transferase
Ciclo dos
nucleotidos da purina
Este ciclo tem
um papel importante no musculo em exercicio
A geração de acido fumarico fornece ao musculo o único substracto
anaplerotico para o ciclo de Krebs
No
decorrer do exercicio as proteinas musculares podem ser utilizadas para
fornecer aspartato
Formação dos
nucleotidotrifosfatos
Resultam
da fosforilação dos mucleotido monofosfatos pelo ATP pela acção de
transfosforilases
Utilização
das purinas livres
As bases puricas resultantes do
metabolismo dos nucleotidos com purinas podem ser recicladas para a síntese de novo dos nucleotidos, por
combinação com o PRPP
A adenina origina o AMP combinando-se
com o PRPP pela acção da adenina
fosforibosiltransferase e a adenosina combinando-se com o ATP pela acção da
adenosina cinase
A hipoxantina e a guanina
combinam-se com o PRPP para dar respectivamente IMP e GMP pela nacção da
hipoxantina-guanina transferase
Regulação da
síntese
A PRPP
sintetase é inibida pelos nucleotidos com purina, em especial o AMP e o GMP por
alteração de conformação
A
amidotransferase é inibida num sitio alosterico pelos adenina nucleotidos (AMP,
ADP, ATP) e noutro sitio pelos guanina nucleotidos
Um excesso de ATP aumenta a
síntese de AMP e a um aumento de GTP e AMP
Capitulo 33
SINTESE DOS NUCLEOTIDOS PIRIMIDICOS
Origem dos carbonos
Os carbonos provêm do acido aspartico,
glutamina e anidrido carbónico
Síntese do UMP
Formação do carbamil-fosfato


{kind=link}
intese do
carbamilaspartato
Forma-se por combinação do carbamilfosfato com o aspartato
pela acção da aspartato transcarbamilase
Formação do acido orotico
O carbamilfosfato desidrata-se em acido dihidroorotico que
por sua vez cede hidrogénio para o NAD para originar o acido orotico.

{kind=link}
Formação do UMP
O acido orotico
combina-se com o fosforibosil pirofosfato dando acido orotidilico que em
seguida origina o UMP por descarboxilação

{kind=link}
Síntese das CMP e TMP
O UMP transforma-se
em UTP que aminado pela glutamina originando o CTP. Este forma o dUMP que em
seguida se metila em TMP
Para formar o CMP o CTP sobre duas desfosforilações
Regulação
Carbamilfosfato sintetase
É a principal via reguladora
É inibida pela UTP, produto final da reacção e activada pelo PRPP
OMP descarboxilase
A UMP compete com o OMP na OMP-descarboxilase
Capitulo 34
CATABOLISMO DAS BASES PURICAS E
PIRIMIDICAS
Bibliografia
Bases puricas
Desaminação da adenina em hipoxantina e da guanina em
xantina pela acção das respectivas desaminases
A hipoxantina é oxidada em
xantina pela hipoxantina-oxidase
A xantina-oxidase oxida a xantina em acido úrico
Bases pirimidicas
O catabolismo das pirimidinas é
complexo
Os produtos finais da degradação são a beta-alanina para o
uracilo e o acido beta-amino butirico
para a timina
Capitulo 35
PATOLOGIA DAS PURINAS
Xantinuria
Causas
Deficiência em xantina-oxidase
Sintomas
Xantina elevada
Acido úrico baixo
Cálculos renais de xantina
Depósitos cristalinos nos rins e músculos esqueléticos
Tratamento
Restrição de alimentos ricos em
purinas
Ingestão de líquidos
Alopurinol – Bloqueia a conversão da hipoxantina em xantina,
esta ultima menos solúvel
Hiperuricemia
Causas sistémicas
Clearance renal de uratos
diminuída, como é o caso do tratamento prolongado com diuréticos
Aumento da sintese de purinas por
um turnover aumentado de nucleoproteinas, que se passa nalgumas situações
hematológicas ( linfomas, leucemias, anemia hemolítica)
Ingestão aumentada de alimentos ricos em purinas, em
especial se forem acompanhados de bebidas alcoólicas
O álcool induz o catabolismo dos nucleotidos no fígado
aumentando a secreção de acido láctico que bloqueia a secreção de uratos pelo
rim
Causas genéticas
Deficiência
em hipoxantina-guanina-fosforibosil-transferase
Hiperactividade a PRPP sintetase
Sintomas
O plasma fica saturado em acido
urino concentrações superiores a 7 mg%
Os uratos são solúveis até 4mg%
A partir destes limites pode
cristalizar e depositar-se em artilagens, tendões e ligamentos
Quando os cristais se agregam
formam tofos visíveis aos R.X.
Se a urina é acida pode formar
cálculos renais
Há casos de hiperuricemia sem
gota
Tratamento
Alopurinol,
análogo estrutural da xantina, que inibe a xantina-oxidase
Os anti-inflamatorios não
esteroides são eficazes no tratamento da fase aguda da gota, mas não baixam a
hiperuricemia
Sindroma de Lesch-Nyhan
Causas
Perda do gene da HRPT
Doença ligada ao sexo pois o gene encontra-se no cromossoma
X
Sintomas
Gota severa
Malfunção grave do sistema nervoso
Tendência para a auto-mutilação
Tratamento
Prevenção da insuficiência renal
Alopurinol
Esforços para reduzir a automutilação
SCID( Doença de Imunodeficiência
severa combinada)
Causas
Deficiência em adenosina
desaminase
Esta deficiência leva à destruição dos linfocitos B e T
Na falta deste enzima, a desoxiadenosina é fosforilada
provocando teores elevados de dATP, inibidor da nucleotido redutase com a
consequente inibição da síntese do DNA
Sintomas
Imunodeficiência
Tratamento
Transfusões
Transplante da medula
Substituição enzimática
Terapia genica
Glicogenose tipo I
A deficiência em glicose-6-fosfatase leva a uma síntese
aumentada de pentose-fosfatos
Há assim mais ribose para formar PRPP
CAPITULO 36
Formação dos desoxiribonucleotidos
A ribonucleotido- redutase catalisa a formação de
desoxiribonucleotidos a partir de nucleotidos
O dador de hidrogénio para a redução da ribose é tioredoxina
reduzida
A tioredoxina é uma proteína com enxofre tendo SH quando reduzida e S-S quando oxidada
A tioredoxina oxidada é reduzida pelo NADPH pela acção da
tiopredoxina redutase
A tioredoxina reduzida cede os seus H para formar os
desoxiribonucleotidos
Regulação
O dATP e dTTP são efectores
alostericos positivos
O ATP acelera a redução dos ATP e CDP
O GTP acelera a redução do ADP e GDP
Capitulo 37
SINTESE DE OUTROS NUCLEOTIDOS
NAD e NADP
Condensação do nicotinato ou da
nicotinamida com o PRPP
Forma-ser o nicotinamida ribonucleotido pela acção da nicotinato ou nicotinamida fosfotransferase
O nicotinamida mononucleotido(NMP) pode formar-se também
pelo metabolismo da triptofana,oque permite formar o NAD e NADP mesmo numa
alimentação pobre em nicotinamida e acido nicotínico
O mononucleotido combina-se com o ATP pela acção da NAD
pirofosforilase para da o NAD
A NAD cinase fosforila o NAD em NADP
FAD
A riboflanina é fosforilada pela
flavocinase para dar ao FMN
Pela acção da FAD
pirofosforilase o FMN liga-se com o AMP para dar o FAD
CoA
O acido pantotenico é fosforilado pelo ATP em 4-fosfopantenato
pela pantenato cinase
Este combina-se com a cisteina para dar
fosfopantenilcisteina, reacção catalisada pela fosfopantenocisteina sintetase
Esta descarboxila-se em 4-fosfopantetina pela acção da
fostopantotenoil descarboxilase
Esta liga-se ao AMP para dar a defosfoCoA pela acção da
defosfoCoA pirofosforilase
A defosfoCoA cinase fosforila o grupo 3’-OH da adenosina
para dar o CoA
Capitulo 38
CATABOLISMO DOS ACIDOS NUCLEICOS E
NUCLEOTIDOS
Ácidos nucleicos
As nucleoproteinas são cindidas pelas proteases em proteínas
e ácidos nucleicos
Os ácidos nucleicos são cindidos por endonucleases e
exonucleases
Endonucleases
Desoxiribonucleases do pâncreas
Cindem ligações 3’-5’ para
produzir grandes nucleotidos 5’-fosfoterminais

http://en.wikipedia.org/wiki/Endonuclease
Desoxiribonucleases do baço
Cindem ligações entre o fosfato e o carbono 5’ produzindo
nucleotidos 3’-fosfoterminais
Ribonuclease
Existem no intestino, fígado e
pâncreas
Hidrolisam ligações fosfato ligadas ao 5’ produzindo
nucleotidos 3’-fosfoterminais
Requerem uma pirimidina no nucleotido 3’-fosfoterminal
Exonucleases
Cindem a ligação fosfodiester na extremidade da cadeia
polinucleotidica
A mais importante é
aa exonuclease do baço que liberta nucleotidos -3-fosfato
´Catabolismo dos nucleotidos e nucleosidos
Os nucleotidos são
desfosforilados por nucleotidases
Os produtos finais são
a pentose-fosfato e a respectiva
base
Sem comentários:
Enviar um comentário