METABOLISMO DOS GLUCIDOS
Digesrão e transporte dos glucidos
Patologia da digestãoa e transporte dos glucidos
Glicolise
Destinos do acido piruvico
Produção de energia do musculo
Patologia da glicolise
Glicogenese
Glicogenolise
Glicogenoses
Neoglicogenese
Regulação do metabolismo dos glucidos
Patologia da neoglicogenese
Regulaçao da glicemia
Diabetes
Sindroma plurimetabolico
Ciclo de Dickens-Horecker
Patologia do ciclo de Dickens- Horecker
Metabolismo das hexoses e da lactose
Patologia do metabolismo das oses
Metabolismo do acido glicuronico
Metabolismo dos derivados das
Patologia da digestãoa e transporte dos glucidos
Glicolise
Destinos do acido piruvico
Produção de energia do musculo
Patologia da glicolise
Glicogenese
Glicogenolise
Glicogenoses
Neoglicogenese
Regulação do metabolismo dos glucidos
Patologia da neoglicogenese
Regulaçao da glicemia
Diabetes
Sindroma plurimetabolico
Ciclo de Dickens-Horecker
Patologia do ciclo de Dickens- Horecker
Metabolismo das hexoses e da lactose
Patologia do metabolismo das oses
Metabolismo do acido glicuronico
Metabolismo dos derivados das
CAPITULO 1
DIGESTÃO E TRANSPORTE DOS GLUCIDOS
Amilases
Amilase salivar
Encontra-se na saliva como
ptialina
A digestão do amido começa na
boca
A mucina salivar é importante
para a sua lubrificação e dispersão
A amilase salivar hidrolisa ao
acaso ligações internas a-1,4 – é
portanto uma endoglicosidase
Quando o bolo alimentar chega ao
estomago, a amilase é desnaturada, deixando de actuar, excepto uma pequena
fracção contida no interior do bolo alimentar



Acção da amilase salivar
Amilase pancreática
O processo digestivo continua quando os alimentos chegam ao
duodeno.
As secreções pancreática contêm bicarbonato e amilase
Os bicarbonatos neutralizam a acidez, permitindo a acção da
amilase pancreática
Nesta fase formm-se diholosidos com ligações 1-4( maltose e
isomaltose) e 1-6 (isomaltose) e oligosidos contendo até 8 residuos glicosilo
alguns com ligações 1,6 (dextrinas-limite)2069
Bibliografia
Glicosidases
A hidrolise dos diholosidos e das dextrinas limites faz-se
através de glicosidases colocadas nas membranas das células em escova das
vilosidades intestinais.
As glicosidades agruparam-se em quatro complexos diferentes:
Complexo sacarase-isomaltase
Hidrolisa a sacarose, maltose e isomaltose
Tem duas subunidades a sacarase
, que hidrolisa a sacarose e a maltose e a isomaltase
Está mais concentrada no jejuno, diminuindo para as
extremidades proximal e distal do intestino
Complexo glico-amilase
Actua sobre as dextrinas limite e é também uma maltase.
A sua actividade aumenta progressivamente ao longo do
intestino, sendo máxima no ileon
Complexo
beta-glicosidase( lactase)
Hidrolisa a lactose em glicose e galactose
A sua distribuição no intestino é igual à do complexo
sacarase-isomaltase
Trealase
Hidrolisa o diholosido trealose
A trealose não é um componente essencial da alimentação pois
encontra-se em insectos, algas e cogumelos.
Foram descritos sintomas graves num deficiente em trealase
que ingeriu muitos cogumelos
Absorção e transporte dos glucidos
Glicose
A glicose é muito polar e não se
difunde através dos fosfolipidos da membrana celular.
Cada oxidrilo da glicose forma
pelo menos duas pontes de hidrogénio com moléculas de agua.
Este objectivos conseguem-se com
proteínas de transporte, designadas por família GLUT
Há dois tipos de transporte -os
dependentes do sódio, utilizando a bomba de sódio, e os de difusão facilitada, independentes do sódio.
Dependentes do sódio
São os transportadores GLUT 5 localizados no lado luminal
das células absortivas intestinais.
Permitem concentrar glicose a partir do lume intestinal
Neste processo o transporte
da glicose está associado ao transporte activo de sódio gerado pela
bomba de sódio, embora a glicose seja captada por difusão facilitada.
A energia dará a captação do sódio é dada pela hidrolise do
ATP, catalisada pela Na+K+ ATPase.
O sódio move-se para o sangue por troca com o potássio


Transporte dependente do sodio
Transporte
independente do sódio
GLUT 1 e 3
Encontram-se em todas as células menos as do fígado e
pâncreas.
São responsáveis pela captação básica de glicose, permitindo
uma difusão lenta mas regular da glicose
GLUT 2
Encontram-se no fígado e células beta do pâncreas
Permitem a entrada de glicose para o fígado e células beta
quando o GLUT 1 está saturado
GLUT4
Encontram-se nos adipocitos e nas células dos músculos esqueléticos
São regulados pela insulina
Quando este transportador está saturado a secreção de
insulina aumenta
A insulina activa o gene GLUT 4
Capitulo 2
PATOLOGIA DA
DIGESTÃO E TRANSPORTE DOS GLUCIDOS
Cólera
A exotoxina do vibrião da cólera inibe a formação do AMP
cíclico, o que acarreta a diminuição da
absorção de agua e sódio do lume pelas células intestinais
Acresce-se o aumento de cloro, catiões e agua no lume, pela
acção da toxina sobre as criptas
Estes factos levam a uma diarreia intensa que pode atingir 1
l./hora com grande perda de solutos, o que acarreta uma desidratação intensa.
Exotoxina
« AMP cíclico
criptas
«
Absorção de Na e K » cloro, catiões e
agua
DIARREIA ATÉ l l/h
Desidratação
Como o transporte dependente de sódio não é afectado pela
toxina, a administração oral de glicose e sódio aumenta a captação de glicose e
sódio, acompanhada da de cloro e água, diminuindo assim a gravidade dos
sintomas
Deficiência em
lactase
Deficiência congénita em lactase
É muito rara
A lactose acumulada transforma-se
em acido láctico provocando diarreias intensas e cólicas após a introdução de
leite ou produtos lácteos
Deficiência em lactase associada à prematuridade
O enzima não é sintetizado na
altura do nascimento mas passa a sê-lo após alguns dias
Deficiência do adulto
A maior parte é adquirida
Muitas alterações generalizadas
do intestino podem produzir deficiências em dissacaridases pois estes enzimas
estão colocados no bordo em escova das células intestinais.
Possivelmente acontece em
indivíduos geneticamente predispostos
Tratamento
Nas crianças deverá ser
prescrita uma alimentação sem leite. A dieta deverá evitar a falta de
nutrientes essenciais
O adulto com esta deficiência tolera pequenas quantidades de
leite e por isso deve conhecer os limites
O iogurte e o queijo são melhor
tolerados devido à fermentação parcial da lactose
Deficiências em sacarase – isomaltase e em
maltase
A deficiência congenita em
sacarase-isomaltase é mais frequente que a da lactase
As deficiências adquiridas de
sacarase-isomaltase e de maltase são muito raras
Os sintomas são semelhantes aos
da insuficiência em lactase
Doença
celiaca
Causas
A mucosa do intestino delgado proximal é sensivel ao glúten
alimentar
A doença não surge sem a ingestão previa de produtos com
glúten
Normalmente surge entre os seis meses e 2 anos e constitui
uma intolerância permanente ao glúten
O glúten encontra-se no trigo, centeio e cevada
Patogenese
A acção deve-se à gliadina que contem sequencias de
aminoacidos repetidas que levam à sensibilização dos linfocitos da lamina
própria
Há uma predisposição genética
A resposta imunitária desencadeia uma reacção inflamatória
que leva à atrofia das vilosidades, hiperplasia das criptas e lesão do epitelio
superficial do intestino delgado
A lesão é mais intensa no intestino delgado proximal e
estende-se progressivamente para a parte distal
Este facto explica a
variabilidade dos sintomas
Sintomas
O mais comum é a diarreia
10% das crianças têm atraso de crescimento sem diarreia
Anorexia e perda de peso são frequentes
Tratamento
Alimentação totalmente isenta de glúten durante toda a vida
Todos os produtos com trigo, centeio e cevada devem ser
proscritos
Muitos alimentos processados têm glúten
Crise celíaca
Diarreia grave, perda de peso, hipocalcemia, hipoproteinemia
O tratamento pode incluir corticoides
Capitulo 3
GLICOLISE
Provem das palavras gregas glyko(doce) e lysis(desaparecimento)
É a transformação da glicose em ácido piruvico ou acido láctico
em anaeróbios
É uma via citoplasmica
Nas leveduras é a via final da destruição da glicose
Nos animais, à glicolise segue-se a via aerobica,
representada pelo ciclo de Krebs
Etapas
A animação seguintes permitem uma boas compreensão da
glicolise:
Formação da
glicose-6- fosfato
A glicose para entrar na célula tem de se fosforilar em glicose-6-fosfato,pela acção da hexocinase
A energia é fornecida pelo ATP


Isoenzimas da
hexocinase
Na maior parte dos tecidos existem os isoenzimas I, II e
III, muito sensíveis mas pouco específicos
No fígado é abundante o enzima IV ou glicocinase enzima especifico mas pouco sensível, o que lhe permite
actuar sobre as altas concentrações de
glicose que se observam após as refeições
As hexocinases são enzimas
alostericos, regulados pelo produto da reacção, a glicose-6-fosfato
Formação da
fructose-6-fosfato
É a conversão duma aldose numa cetose
O enzima é a fosfohexose
isomerase

http://www.dbio.uevora.pt/jaraujo/biocel/glicolise.htm
Formação da fructose
– 1,6 – bisfosfato
A fosfofructocinase
catalisa a fosforilação da frutose-6- fosfato


É uma reacção irreversivei, necessitando de ATP
Criou-se um ponto fraco na molécula que irá facilitar a sua
posterior cisão

Cisão da frutose-1,6-
bis fosfato
A frutose 1-6 bisfosfato
é cindida em duas triose-fosfato, o fosfogliceraldedo e a
fosfodihidroxiacetona pela acção da aldolase
As duas triose fosfato isomerizam-se uma na outra pela
triose-fosfato isomerase
Como o fosfogliceraldeido é a única triose a ser utilizada,
a fosfodihidroxiacetona ir-se-á transformando em fosfogliceraldeido

Formação do
gliceraldeido 1,3-bisfosfato
A triose fosfato deidrogenase desidrogena o gliceraldeido
1,3 – bisfosfato, associada a uma fosforilação, com a formação de uma ligação
fosfato de forte potencial
O hidrogénio libertado é captado pelo NAD


Transformação em
3-fosfoglicerato
O fosfato de forte potencial é transferido para um ADP,
formando-se 3 – fosfoglicerato.
A reacção é catalisada pela fosfogliceratocinase


Isomerisação em
2-fosfoglicerato
Esta reacção é catalisada pela fosfogliceromutase
Formação do acido
fosfoenolpiruvico
A enolase catalisa a formação de um enol e de um fosfato de
forte potencial
Formação do acido
piruvico
Forma-se por desfosforilação pela acção da piruvato cinase
com formação de um ATP



Regulação da glicolise
Fosfofructocinase
É o enzima regulador primário da glicolise
É inibida alostericamente pelo ATP que diminui a sua
afinidade para o substracto
O citrato é um inibidor porque impede a ligação do ATP ao
sitio alosterico
Quando o estado energético da célula é baixo (AMP e ADP
baixos), o ATP é deslocado do sitio
alosterico pelo AMP
Fructose-2,6 –
bisfosfato
É o efector alosterico mais potente da fosfofrutocinase
A sua síntese é catalisada por um enzima bifuncional, a fosfofrutocinase 2, que actua como
fosfatase quando fosforilada e como cinase como desfosforilada
A fosforilação é activada pelo AMP cíclico. Por esta razão
os estimuladores do AMP cíclico (
adrenalina, glucagina) inibem a glicolise e os inibidores(insulina) estimulam

Hexocinase
As hexoquinases I,II e III, mas não as IV são reguladas
indirectamente pela fosfofrutocinase 1.
Se este enzima é inibido acumula-se frutose-6-fosfato que
pela lei da acção das massas forçará a reacção reversível da
fosfoglicoisomerase, originando a glicose-6-fosfato que é um inibidor das hexocinases
Piruvato-cinase
Tem quatro subunidades
Tem três isoenzimas : M
existente no musculo, L no fígado e A nos outros tecidos
Pode haver duas formas de regulação: alosterica e
fosforilação
Regulação alosterica
O substrato liga-se às quatro subunidades do enzima de um modo
cooperativo, de um modo semelhante à ligação do oxigénio com a hemoglobina
Todos os isoenzimas são inibidos pelo acetil-CoA
A fructose-1,6 – bisfosfato é um activador
O ATP e a alanina são inibidores alostericos
Regulação por
fosforilação
Só se passa no isoenzima L
A fosforilação estimulada pelo AMP cíclico inibe o isoenzima
A inibição produzida pelo AMP cíclica, produzida por exemplo
pela glucagina, faz dirigir o fosfoenolpiruvato para a neoglicogenese
Balanço energético
O numero de ATP formados em anaerobiose é reduzido , em
oposição aos formados em aerobiose
Ciclo de
Rapaport-Luebring
No eritrocito encontra-se o ácido 2,3 – bisfosfoglicerico
As suas cargas negativas unem-se às cadeias de carga positiva
da hemoglobina facilitando a expulsão de
oxigénio para os tecidos
Forma-se a partir do acido 1,3-bisfosfoglicerico pelo ciclo
de Rapaport-Luebring
Aumenta em populações vivendo em altas altitudes
devido à falta de oxigénio, em situações de anoxia,e em doenças crónicas em que
haja má distribuição de oxigénio e em anemias graves

CAPITULO 4
DESTINOS DO ACIDO PIRUVICO
Aerobiose
Mecanismos
O acido piruvico é descarboxilado em acetil-CoA
Esta descarboxilação tem um mecanismo semelhante à do acido a-cetoglutarico no ciclo de Krebs
São cofactores o pirofosfato de tiamina(PTT), coenzima A e
acido lipoico
O complexo multienzimatico tem três enzimas – piruvato
deidrogenase( PDH), dihidrolipoil transacetilase e piruvato deidrogenase
Regulação
A piruvato deidrogenase existe sob uma forma activa,
desfosforilada, e inactiva fosforilada
A fosforilação e desfosforilação dependem da PDH cinase e da
PDH fosfatase, respectivamente
Estes enzimas dependem do AMP cíclico
Quando o nível energetico da célula é alto( aumento de
ATP,NADH e acetil CoA)a cinase é activada
0
Anaerobiose
Revisões de conjunto
Fermentação láctica
No decorrer de
esforços musculares intensos, não há um aporte de oxigénio suficiente para
regenerar o NADH
Nesta situação, os hidrogénios
são cedidos ao acido piruvico para formar o acido láctico
O enzima é a lactico deidrogenase


Nos tecidos sem mitocondrias,
como os eritrocitos, o metabolismo do piruvato é sempre anaeróbico.
A acção da láctico deidrogenase é
reversível e assim o acido láctico captado por outros tecidos pode ser
convertido em piruvico. É .o que se passa no musculo cardíaco e alguns músculos
esqueleticos
Fermentação alcoólica
Nas leveduras não há piruvico deidrogenase.
O enzima que actuará sobre o acido piruvico é a piruvico-descarboxilase que descarboxila o acido piruvico em
acetaldeido
O NADH não
regenerado pelo oxigénio, é transferido
para o acetaldeido, formando-se álcool
etílico.
O enzima é a álcool
deidrogenase

.


Neoglicogenese
.
Na gliconeogenese, a formação da
glicose pode começar a partir da carboxilação do acido pirúvico


Destinos do acido piruvico
Capitulo
5
PRODUÇÃO DE ENERGIA NO MUSCULO
Hidrolise do ATP
A forma mais fácil e
eficaz de o musculo obter energia é a hidrolise do ATP
Resintese do ATP
Quando no decorrer de um esforço muscular prolongado, o ATP
terá que ser resintetizado, pois é a única forma de energia utilizada pelo
musculo.
Nestas situações, o musculo recorre a uma reserva
energética, a fosfocreatina, que tem um fosfato de alto potencial
A creatinafosfocinase
hidrolisa a fosfocreatina, libertando o fosfato de alto potencial que irá
resintetizar o ATP
Resintese do ATP e da
fosfocreatina
Findo o esforço, é necessário refazer as reservas
energéticas
É necessário recorrer à glicolise e à fase aeróbia para
formar ATP
Como a glicose foi toda gasta, é
necessário recorrer
à glicogenolise para obter nova glicose.
Os primeiros ATP formados serão utilizados para regenerar a
fosfocreationa
Capitulo
6
PATOLOGIA DA GLICOLISE
Revisões de conjunto
Piruvato cinase
Os eritrocitos maduros dependem totalmente da glicolise para
formar ATP pois não tendo mitocondrias não há vias aerobicas
Na ausência hereditária de piruvato cinase não se forma ATP
As bombas iónicas ATP dependentes não funcionam
Os eritrocitos deixam de ter uma forma bicôncava que lhes
permitia escorregar através dos capilares para fornecer oxigénio aos tecidos
Os eritrocitos tumefazem-se e lisam deixando sair a
hemoglobina, havendo uma anemia que por haver hemolise se chama hemolitica
Hiperlactacidemia
A hiperlactacidemia do recemnascido e da criança jovem
resulta da falta de oxidação do NADH que acarreta haver menos NAD para
transportar hidrogénio
Parte do hidrogénio não transportado combina-se com o acido
piruvico para dar acido láctico
Outras causas possíveis são a falta dos enzimas da
neoglicogenese ou dos enzimas que metabolizam o acido piruvico
Capitulo 7
GLICOGENESE
Dà-se o nome de glicogénese à formação do glicogénio a
partir da glicose.
É um processo que ocorre pràticamente em todos os órgãos e
tecidos, mas principalmente no fígado e músculo
Revisões de conjunto
Principais
etapas
Formação de UDP-glicose
Fosforilação da glicose
em glicose-6-fosfato pela acção de uma hexocinase.
Isomerização desta em
glicose-1- fosfato pela fosfoglicomutase na presença da glicose-1,6-bisfosfato.
A glicose-1- fosfato combina-se com a UDP para dar
UDPGlicose (UDPG) pela acção da UDPG pirofosforilase
Este enzima é inibido competitivamente pela
galactose-1-fosfato, facto que é responsável pela toxicidade da galactose-1-.fosfato
que se acumula na galactosemia

Formação de cadeias lineares
A formação das cadeias lineares faz-se pela acção da glicogénio-sintase
na presença de uma matriz de
glicogénio.
Este enzima transfere
um C1 da glicose UDPG para o C4 de uma glicose terminal, com
libertação de UDP produzindo o alargamento das cadeias 1-4
Esta reacção
repete-se as vezes necessárias



Este alongamento das cadeias necessita de uma proteina
incluida na molécula do glicogénio, a glicogenina que tem a propriedade
de catalisar a sua própria glicosilação ligando o C1 duma UDPG a um resíduo
tirosina.
Formas da glicogénio sintase
Existe sob duas formas – forma a activa, não fosforilada e
forma b, inactiva, fosforilada
Forma
a
A forma a é fosforilada por uma cinase
A cinase tem uma forma inactiva (I ) e uma forma activa (A)
.
A forma I é activada pelo AMP cíclico formado a partir do
ATP por um sistema em cascata activado pela adrenalina
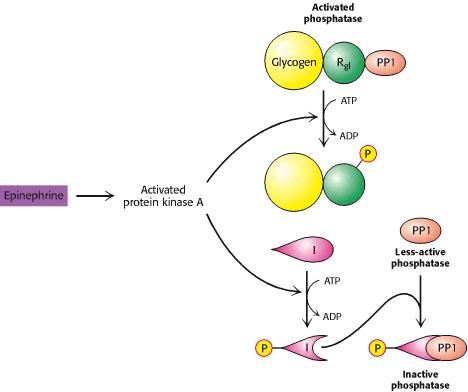
No músculo existe uma outra cinase dependente do cálcio e fosfolípidos, activada pelo diacilglicerol.
É a proteina cinase C.
No musculo pode ser activada por um factor proteico, na
presença de calcio
Existe uma cinase activada pelo sistema cálcio- calmodulina
Forma b
A forma b é
desfosforilada por uma proteina fosfatase I inibida pelo glicogénio e
activada pela insulina e corticosteroides
Regulação das fosforilases
O AMP libertado pela destruição do ATP durante a contracção
muscular activa alostericamente a fosforilase b
Os impulsos nervosos libertam cálcio do retículo
sarcoplasmico
O cálcio liga-se à calmodulina, modificador da cinase
A fosforilato cinase também é activada pela fosforilação da
proteína cinase A, permitindo a ligação da adrenalina aos receptores da
membrana
Introdução de ramificações
A glicogénio sintase forma apenas cadeias lineares (
ligações 1-4 ).
A formação de
ramificações (ligações 1,4-1,6)
necessita do enzima ramificante ou amilo(1,4-1,6)
transglicosidase.
Este enzima transfere
um mínimo de seis glicosilos para um oxidrilo em C6, estabelecendo um ponto de
ramificação

A ramificação cresce em seguida por acréscimo de ligações
1-4 até se fazer outra ramificação
Capitulo 9
GLICOGENOLISE
É o processo pelo qual o glicogénio é degradado em glicose
A glicose formada ou é degradada
para formar ATP ou entra na circulaçãoPode fazer-se no citoplasma ou nos
lisossomas
Glicogenólise
no citoplasma
Degradação das
cadeias lineares
A degradação das cadeias lineares faz-se pela acção de transglicosidases
geralmente conhecidas por fosforilases.
Estes enzimas cindem o ortofosfato, fixando-se o fosfato na
molécula de glicose que se irá cindir e o OH na molécula distal,
formando-se glicose-1-fosfato e uma molécula de glicogénio com menos uma
glicose

Acção sobre as ramificações
.
A reacção vai continuando até surgir a primeira ramificação
1~6 onde irá actuar o enzima desramificante ou amilo-1,6-glicosidase


Fosforilase muscular
Estrutura
A fosforilase a é constituida por quatro subunidades contendo
cada uma uma molécula de fosfato de piridoxal e uma de fosfoserina.
A fosfatase b é
dimerica

Conversão da a em b
A forma a converte-se na b pela desfosforilação das quatro
fosfoserinas pela acção da fosfatase da fosforilase, separando-se em
dois dímeros
É activada pelo AMP e inibida pela glicose-6-fosfato e pelo
ATP
Conversão da b em a
A forma b converte-se em a pela acção da fosforilase
cinase na presença de ATP e
magnésio.
Esta cinase é a mesma que intervem na glicogénese, e tem o mesmo sistema de activação
Fosforilase hepática
As fosforilases hepáticas são diméricas diferindo apenas na
existência de serina ou de fosfoserina.
O sistema é activado pela adenilciclase mas não pelo cálcio.
Formação da
glicose-6- fosfato
Na glicogenolise forma-se
glicose-1- fosfato
Como a forma activa
da glicose é a glicose-6-fosfato, esta terá que se converter pela acção da fosfoglicomutase

Transformação da glicose-6-fosfato em glicose
É uma hidrólise
catalisada pela glicose-6- fosfatase
Passa-se no fígado,
rim e intestino, mas não no musculo
O enzima é reprimido
pela insulina e induzido pelos glicocorticoides

Glicogenólise nos
lisossomas
Nos lisossomas a cisão do glicogénio faz-se por hidrólise e
não por fosforólise., pela acção de uma maltase acida
Passa-se no fígado,
rim e intestino, mas não no musculo
O enzima é reprimido
pela insulina e induzido pelos glicocorticoides
Capitulo 10
Glicogenoses
Devem-se à falta de
um enzima do metabolismo do glicogénio
Number
|
Enzyme deficiency
|
Eponym
|
Symptoms[2]
|
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
|
|||
GSD type VIII
|
(In the
past, considered a distinct condition.[3] Now classified with VI.[4] Has been described as X-linked
recessive.[5])
|
||
-
|
|
||
GSD type X
|
|||
-
|
|
Glicogenose tipo 0
Falta a glicogénio
sintase
Como não se forma
glicogénio, não haverá glicogenolise e por esta razão os doentes não mantêm a glicose em jejum.
Estão indicadas
refeições frequentes ricas em proteínas e à noite suplementos de anido de milho
cru
Glicogenose tipo Ia ou doença de von Gierke
Deve-se à falta de
glicose 6 fosfatase
A glicose-6-fosfato
acumulada inibe a fosforilase e activa a sintase
O glicogénio
acumula-se no fígado e no rim provocando hepatomegalia e insuficiência renal.
Os sintomas surgem
nos primeiros anos de vida com hepatomegalia, hipoglicemia e acidose láctica
O tratamento visa
essencialmente a manutenção da glicemia pela infusão nasogastrica de glicose ou
administração oral de milho cru
Revisões de conjunto
Glicogenose tipo II ou doença de Pompe
Falta a maltase
acida
Acumula-se
glicogénio nos lisosomas
Há hipoglicemia devido à cisão deficiente do glicogénio
Afecta gravemente os
músculos


Glicogenose tipo III, doença de Forbes ou doença dos Cori
Falta o enzima desramificante, formando-se um glicogénio
anormal com muitas cadeias 1-6
Pode observar-se uma
miopatia progressiva
Glicogenose tipo IV ou doença de Andersen
Falta o enzima
desramificante
Acumula-se um
glicogénio com poucas ramificações
O glicogénio pode
comportar-se no fígado como um corpo estranho, desencadeando uma reacção
auto-imune
Leva a uma cirrose
progressiva, insuficiência hepática e morte
Há uma forma
neuromuscular
Glicogenose tipo V ou doença de Mc Ardle
Falta a fosforilase
muscular
Diminui a glicogenolise, acarretando menor produção
de ATP que se reflectirá no exercício físico


Glicogenose tipo VI ou doença de Hers
Falta a fosforilase
hepática
Os principais sinais
são hepatomegalia, hipoglicemia ligeira e acidose
Melhora com a idade
Glicogenose tipo VII ou doença de Tarui
Falta a
fosfofrutocinase
A glicose-6-fosfato
acumulada estimula a síntese da UDP-glicose fosforilase, acumulando-se
glicogénio no musculo
Glicogenose tipo VIII
Falta a cinase da
fosforilase
Acumula-se
glicogénio normal
Capitulo 11
NEOGLICOGENESE
Na ausência de glicose, a manutenção da glicémia faz-se a
partir de precursores não glucídicos.
Este mecanismo de homeostase é indispensável pois o sistema
nervoso central dos mamíferos utiliza apenas a glicose como fonte de energia.
Define-se neoglicogénese como a formação de glicose a partir de
material não glucídico e do lactato.
Todos os precursores vão ser encaminhados para uma via
comum a partir do piruvato ou do lactato
para onde convergem vias especiais indo dos precursores ao piruvato0
Denominam-se sítios de neoglicogénese aos orgãos onde
se realiza a via final. São eles o fígado e o rim e em menor extensão as
células epiteliais do intestino delgado.
A capacidade neoglicogenica destes órgãos contrasta com a
sua fraca capacidade glicolítica
O consumo energético da neoglicogénese é elevado – a
conversão de dois moles de piruvato em glicose exige 4 ATP, 2 GTP e 2 NADH.
Vias especiais
Amino-ácidos
Faz-se a partir dos aminoácidos glicoformadores que por
desaminação ou transaminação originam ácido pirúvico, a-cetoglutárico, oxaloacético ou
acetil-coenzima A
Estes aminoácidos são:
Originando alfa-cetoglutarico
- Acido glutamico
- Histidina
- Hidroxiprolina
- Arginina
Originando acido
oxaloacetico
- Acido aspártico
Originando acido
piruvico
- Alanina
- Serina
- Glicocola(via serina)
- Treonina (via glicocola)
- Serina
- Cisteina
Originando
succinil-CoA(via propionil-CoA)
- Treonina
- Valina
- Isoleucina
Lípidos
Acidos gordos com um numero par de carbonos
Por beta-oxidação formam acetil-CoA
Alem disso altos níveis de acetil CoA activam a piruvico
carboxilase e inibem a piruvico deidrogenase
Ácidos gordos com um numero impar de carbonos
Formam propionil-CoA que por sua vez origina o succinil-CoA

Conversion of Propionyl-CoA to
Succinyl-CoA
Via
final
A neoglicogénese não é o inverso da glicólise porque nem
todas as reacções reversíveis devido a barreiras energéticas.
Os enzimas que funcionam numa só direcção são os enzimas
chave( glicolíticos ou glicogénicos).
Os enzimas que
funcionam nas duas direcções são os enzimas bifuncionais.
Enzimas glicoliticos chave
Barreiras
energéticas impedem que algumas reacções sejam reversíveis .São as reacções
catalisadas por
- Hexocinases
- Fosfofrutocinase 1
- Piruvato cinase
Enzimas glicogenico chave
São enzimas que
catalisam as reacções inversas dos enzimas glicogenicos chave:
¨
Formação do ácido enolpirúvico
¨
Desforilação da fructose –1,6- bisfosfato em
frutose-6-fosfato
¨
Passagem de glicose-6- fosfato a glicose
Formação do ácido fosfoenolpirúvico
Esta reacção faz-se em duas étapas:
¨
Formação do ácido oxaloacético
¨
Descarboxilação fosforilante deste em ácido
fosfoenolpirúvico

Formação de ácido oxaloacético
Trata-se da reacção de Wood e Werkman, catalisada pela piruvato carboxilase.
Necessita de ATP.
Tem a biotina como coenzima.
A fixação da biotina ao enzima é activada pelo acetil-CoA
CO2
ATP
Acido pirúvico --------------- Acido oxaloacetico
Piruvico carboxilase
Descarboxilação fosforilante do ácido
oxaloacético
O fosfato de alto potencial necessário para esta reacção vem
do GTP
.O enzima que catalisa esta reacção é a fosfoenolpiruvato carboxicinase

cortesia de Joyce Diwan
Travessia das mitocondrias
O ácido oxaloacético forma-se nas mitocôndrias mas a sua
transformação em fosfoenolpirúvico é extramitocondrial.
Como o oxaloacético não atravessa a membrana mitocondrial,
terá que se transformar num composto que atravesse a membrana e em seguida se
reconverta nele.
Há duas vias – a via
do malato e a via do aspartato.
Via do malato
O oxaloacetato é reduzido em malato pela malatodeidrogenase
mitocondrial na presença de NADH.
A translocase dos ácidos dicarboxílicos faz o malato
abandonar as mitocôndrias para este no citoplasma dar de novo oxaloacetato.
Via do aspartato
A aspartatoaminotransferase transamina o oxaloacético em
ácido aspártico.
Uma translocase transfere o
ácio aspártico para o citoplasma
onde se converte de novo em oxaloacético

Desfosforilação da frutose-1,6-bisfosfato
A frutose-1,6-bisfosfatase catalisa esta reacção.
O ADP é um efector alostérico negativo ao mesmo tempo que
activa a fosfofrutocinase.
O enzima é activado pelo ácido láctico e pelo cortisol.
Desfosforilação da glicose-6-fosfato
A hidrólise irreversível do fosfato é catalisada pela
glicose-6-fosfatase.
O enzima está fortemente ligado à membrana
A glicose-6-fosfato é transportada para o retículo por um transportador, sendo
aí hidrolisada
A glicose formada é transportada de novo para o citossol por
um transportador
Necessita de
fosfolípidos para a sua actividade.
É inibido pelos fosfatos e pela glicose
Necessita de três transportadores
1. Transporte
d a glicose-6-fosfato para o lume
2. Transporte
do fosfato para o citossol
3. Transporte
da glicose para o citossol
Ciclo dos Cori
O músculo não consegue
transformar em piruvato o ácido láctico formado após um esforço muscular
intenso.
Para se converter em piruvato, o ácido láctico tem que
ser transportado para o fígado ou rim,
que têm enzimas que permitem fazer esta conversão- é o ciclo dos Cori

Ciclo de Fehlig
Uma outra forma de
metabolização do acido piruvico
pelo musculo para fins glicogenicos é a sai conversão em alanina
A alanina
dirigir-se-á para o fígado, onde será reconvertida em acido piruvico
É o ciclo de
Fehlig


Regulação
A glicolise e a neoglicogenese são controladas pelos mesmos
mecanismos, para que funcione apenas uma das vias
A fosfofrutocinase 1 é
estimulada pelo AMP e inibida pelo ATP e citratos, efectores que têm uma acção
oposta sobre a frutose-1,6-bisfosfatase
Assim, quando há um baixo nível energético, a glicolise é
estimulada e no caso contrario é estimulada a gliconeogenese
A frutose-1,6-
bisfosfato tem níveis baixos na inanição e elevados na saciedade
O antagonismo glucagina-insulina implica a estimulação da
gliconeogenese na inanição e da glicolise na saciedade
A piruvatocinase
é inibida pela ATP e Alina, ao contrario da carboxicinase
Capitulo 12
REGULAÇÃO DO METABOLISMO DO GLICOGENIO
A regulação do metabolismo do glicogénio faz-se através de
dois enzimas fundamentais, a glicogénio sintase e a fosforilase.
O AMP cíclico desempenha um papel fundamental na regulação
destes enzimas pois mediante a fosforilação destes enzimas inibe a sintase e
estimula a fosforilase actuando assim no
sentido da glicogenólise.
AMP cíclico
O AMP cíclico actua no metabolismo do glicogénio no sentido
da glicogenolise pois fosforila a forma a da glicogénio sintetase, tornando-a
inactiva e activa a forma a da fosforilase

RESUMINDO:
GLUCAGINA ( fosforilação)
Fosfatase a
Sintase b
GLICOLISE
INSULINA ( desfosforilação)
Fosfatase b
Sintase a
GLICOGENESE

Inibidor proteico I
A glicogenólise é uma forma rápida de mobilização da glicose
que só deverá funcionar quando for necessário mobilizà-la ràpidamente.
Esta regulação faz-se pelo balanço
fosforilação-desfosforilação, encaminhando a fosforilação para a glicogenólise
e a desfosforilação para a glicogénese.
A desfosforilação é assegurada pela fosfoproteina fosfatase
I que desfosforila a glicogénio sintase, a fosforilase e a cinase.
Este enzima é inibido pelo inibidor proteico I que se
forma por fosforilação da sua forma inactiva pela acção da proteina cinase
formada pelo AMP cíclico.
O inibidor é inactivado ao ser desfosforilado por uma
fosfoproteina fosfatase I.
RESUMINDO: Ao ser activado o inibidor proteico I activa
todos os enzimas que levam à glicogenólise passando-se o contrário quando é
desfosforilado.
Capitulo 13
PATOLOGIA DA NEOGLICOGENESE
Deficiência em Glicose-6-fosfatase
É a glicogenose tipo I
Deficiência em
frutose-1,6-bisfosfatase
Sintomas
Hiperventilação
Convulsões
Coma
Tratamento
Tratamento das crises
agudas pela infusão intravenosa de glicose
Evitar jejum
Restrição de frutose e sacarose
Para prevenção da hipoglicemia dar um glucido de libertação
lenta como o amido de milho
Deficiência em
piruvico carboxilase
Consequência da
deficiencia
Deplecção de oxaloacetato
acarretando níveis reduzidos de aspartato, metabolito necessário para a síntese
da ureia
Sintomas
Acidose láctica
Hiperamoniemia
Tratamento
Suplementos de aspartato e citrato
Capitulo 14
Regulação da glicemia
O fígado dos mamíferos é capaz de responder a níveis
diferentes de glicose circulante. Quando o teor em glicose do sangue portal é
alto, o fígado absorve mais glicose. Quando é baixo, liberta glicose.
Como as células hepaticas são totalmente permeáveis à
glicose, pensa-se que o balanço entre a absorção e a libertação de glicose se
deve à actividade dos enzimas glicoliticos e glicogenicos chave.
Na regulação da glicemia as hormonas desempenham um papel
importante.
AMP cíclico
Muitas hormonas actuam sobre o AMP cíclico
Insulina
A insulina inibe a formação do AMP cíclico
Combina-se com um receptor que induz a síntese de um segundo
mensageiro que inactiva a cinase
Induz os enzimas glicolitico chave inibe os glicogenico chave
Induz os enzimas da
lipogenese e inibe os lipoliticos ( acção sobre o AMP cíclico)
Alem disso facilita a entrada da glicose na celula
Favorece a
glicogenese



{kind=link}
Adrenalina
A adrenalina estimula a produção de AMP cíclico
combinando-se com uma proteina específica existente no interior da membrana, o receptor
adrenérgico.
O complexo receptor- adrenalina na presença de ATP
combina-se com a proteina G estimulando
a adenilciclase.
A adrenalina actua apenas no músculo e não no fígado

Glucagina
No fígado a glucagina toma o lugar da adrenalina.
Como resposta a uma descida da glicose sanguínea as células
do pâncreas segregam glucagina que combinando-se a um receptor estimula a
adenilciclase por um mecanismo semelhantre ao da adrenalina, favorecendo assim
a glicogenólise

Na figura seguinte estão esquematizadas as acções da
insulina e glucagina na regulação da glicemia

{kind=link}
Glicocorticoides
- Activam
o catabolismo das proteínas e lipidos, favorecendo a neoglicogenese
- O
acetil CoA formadono catabolismo
dos lipidos activa a piruvato carboxilase, enzima glicogenico chave, e
inibe os glicoliticos chave
- Induzem
a síntese dos enzimas glicogenico chave
Capitulo 15
DIABETES
Conceitos
A diabetes mellitus é uma doença heterogenica caracterizada
pela presença de hiperglicemia
A hiperglicemia é
devida a uma deficiência da acção da insulina
As duas principais causas são a menor produção de insulina
pelo pâncreas ou uma resposta deficiente
da insulina nos órgãos alvo
Estas duas causas definem dois tipos de diabetes – tipo 1 e
tipo 2 respectivamente
Diabetes tipo 1
Causas
É causada pela destruição autoimune das células beta dos
ilhéus de Langerhans
A reacção inicia-se por um mecanismo desconhecido
A destruição das células beta desencadeia uma menor produção
de insulina com hiperglicemia e os outros sinais de diabetes
Parece ser o resultado da combinação de uma susceptibilidade
genetica com factores ambientais
Fases
Existência de autoanticorpos com glicose pós-prandial normal
Diminuição da tolerância à glicose
Hiperglicemia em jejum embora se produza ainda insulina
suficiente para produzir a cetose
A produção de insulina desce ainda mais e os doentes
tornam-se dependentes da insulina exógena
Complicações
São a aterosclerose, neuropatia periférica, insuficiência
renal, retinopatia
O desenvolvimento e gravidade destas complicações dependem
do substracto genético e do grau de controle metabólico
Um controle rigoroso da glicemia reduz o risco das
complicações de 35 a 75%
Diabetes tipo 2
Causas
Surge no adulto, quase sempres após a meia idade
Não é autoimune
A susceptibilidade genética é um requisito indispensável mas
a sua expressão clínica está determinada em parte por factores ambientais
Nos tipo 2 a resposta à ingestão de glicose é inadequada e
os padrões basais são elevados, sinais de resistência à insulina
Complicações
As mesmas do tipo 1
Tratamento
A perda de peso, o aumento do exercício físico e a supressão
do açucar são os métodos mais efectivos
Quando não resultam totalmente devem ser complementados com
hipoglicemiantes orais como as sulfanilureias ou as biguanidas
Capitulo 16
Capitulo 17
CICLO DE DICKENS-HORECKER
Conceito
Via que utiliza os seis carbonos da glicose para gerar
equivalentes redutores e pentoses
Também é conhecido
por ciclo das pentoses, via das pentose-fosfatos e desvio das hexose-fosfatos
Realiza-se no figado, tecido adiposo, córtex suprarenal, testículos, glândula mamaria
lactante e eritrocitos
Tem uma fase oxidativa e uma não oxidativa
Via oxidativa


Formação de
fosfoglicolactona
O C1 da glicose-6-fosfato perde dois hidrogénios, captados
pelo NADP
A reacção é catalisada pela glicose-6-fosfato deidrogenase
ou enzima intermediário de Warburg
É inibido por certas drogas como as sulfonamidas e a
primaquina e activada pela insulina
Mais de 100.000 pessoas têm uma deficiência hereditária neste enzima
Formação de acido fosfogluconico
A lactonase transforma a fosfoglicolactona em acido
fosfogluconico

Formação de
ribulose-5-fosfato
Forma-se uma cetona intermediaria, sendo os hidrogenios
captados pelo NADP
A cetona intermediaria é descarboxilada em
ribulose-5-fosfato

O NADPH formado é utilizado no metabolismo dos lípidos
Isomerização das
pentoses-fosfato
A fosfopentose-epimerase
transforma a ribulose-5-fosfato em xilulose-5-fosfato e a fosfopentose
isomerase isomeiza-a em ribose-5- fosfato

Há uma série de
transferências de dois e três carbonos com formação de trioses e hexoses

1ª transcetolização
Transfere o C1 e C2 (cetol) da
xilulose-5- fosfato para a ribose-5-fosfato originando uma cetose, a
sedoheptulose-7-fosfato e o gliceraldeido-3-fosfato
A reacção é catalisada pela
transcetolase
Dois compostos com cinco carbonos,
originaram um com 7 e um com 3

Aldolização
A aldolase catalisa a
transferência de três carbonos da sedoheptulose-7-fosfato para o fosfogliceraldeido
para originar um composto com quatro carbonos (eritrose-4-fosfato) e um com
seis (frutose-6-fosfato)

2ª transcetolização
A transcetolase efectua uma nova
recombinação os dois primeiros carbonos da xilulose para a eritrose
Forma-se frutose-6-fosfato e
fosfogliceraldeido


{kind=link}
{kind=link}
Equilentes redutores sob a forma de NADPH que serão utilizados em reacções de
síntese
Fornecer ribose
para a síntese dos ácidos nucleicos
Manter
a integridade da membrana dos eritrocitos pela redução do glutatião
Metabolisar
as pentoses alimentares
Regulação
O factor mais
importante é a concentração celular de
NADPH
A
disponibilidade em NADP regula a reacção limitante, a reacção da
glicose-6-fosfato-deidrogenase
Capitulo 18
PATOLOGIA DO CICLO DE DICKENS-HORECKER
´
Glutatião
O glutatião tem uma actividade anti-oxidante por destruír os
peróxidos
A regeneração do glutatião faz-se pela acção da glutatião
peroxidase, que necessita de NADPH
Como o ciclo de Dickens.Horecker é o principal fornecedor de
NADPH,qualquer falha deste ciclo reflectir-se-á na regeneração do glutatião e
consequente aumento do stress oxidativo
Eritrocitos
Nos eritrocitos a única fonte de NADPH é o ciclo de
Dickens-Horecker
Qualquer diminuição de produção de NADPH por este ciclo terá
sempre consequências nefastas, por falta de alternativas
As consequências serão hemolise por enfraquecimento da
parede celular e maior oxidação da hemoglobina em metahemoglobina
Deficiencia em
glicose-6-fosfato-deidrogenase
É a deficiência hereditária mais espalhada no mundo – afecta
mais de 400 milhões
Na bacia mediterrânica e em Africa está muitas vezes relacionada
com a resistência ao Plasmodio falciparum
Nas áreas em que a malária é endémica a deficiência tem uma
prevalência de 5 a 25% enquanto que em áreas não endémicas é inferior a 0,5%
A causa dos sintomas é a falta de produção de NADPH
Esta deficiência se manifesta por anemia hemolítica
Manifesta-se na presença de oxidantes
Também tem o nome de favismo por aparecer após a ingestão de
favas, que é um oxidante.
Alimentos e
medicamentos a evitar
Há alimentos e medicamentos que devem ser completamente proscritos
para impedir o aparecimento de uma nova crise
Capitulo 19
METABOLISMO DAS
HEXOSES E DA LACTOSE
A quase totalidade do metabolismo das oses e oligosidos
faz-se através da glicose
As outras hexoses podem-se converter em glicose ou em
intermediários da glicose, do mesmo modo que a glicose pode originar algumas
hexoses
Frutose
É fosforilada pela frutocinase em frutose-1-fosfato.
A frutose-1-fosfato é
cindida pela aldolase da frutose-1-fosfato em gliceraldeido e
fosfodihidroxiacetona
A triose-cinase fosforila o gliceraldeido em
fosfogliceraldeido

Galactose
Embora a galactose
seja um epimero da glicose, a sua
transformação nesta é complexa.
Primeiramente forma-se por accão da galactocinase em
galactose-1-fosfato pela acção da galactocinase
Esta transforma-se em UDP galactose pela acção da
galactose-1-fosfato uridiltransferase
A epimerização faz-se a este nível pela acção da
UDP-galactose-4-epimerase que em seguida irá originar a glicose-1-fosfato

Manose
A hexocinase converte-a em
manose-6-fosfato
A manose-6-fosfato-isomerase converte-a em frutose-6-fosfato
Síntese da lactose
É sintetisada pelo sistema
lactose-sintetase que contem duas proteínas:
Proteína A
Encontra-se não só na glândula mamaria mas também no fígado
e intestino
Forma acetillactosamina pela combinação da UDPGal com a
acetilglicosamina
Proteína B
É sintetisada apenas durante a
lactação
Modula a proteína A para a
formação de lactose
.
Capitulo 20
PATOLOGIA DO METABOLISMO DAS OSES
Galactose
Existem três formas de galactosemia:
- Falta
de Gal-1-fosfato-Uridil transferase
- Falta
de galactocinase
- Falta
de UDP-gal-4-epimerase
Como as duas primeiras têm os mesmos sintomas serão estudadas
em conjunto
Deficiência em
transferase
Causas
O recemnascido ingere 20% das suas calorias como lactose,
que contem galactose
Na falta deste enzima, a
galactose-1-fosfato não se metabolisa, acumulando-se nos rins, fígado e cérebro
A sua transformação em galactitol pode produzir
cataratas
Sintomas
Os recemnascidos ou lactentes
apresentam entre outros sintomas alterações hepáticas, , convulsões ou
letargia, atraso mental e cataratas
Quando o diagnostico não é feito
precocemente a cirrose, o atraso mental e as cataratas tornam-se irreversiveis
Cataratas
É um dos sintomas mais frequentes
Deve-se à conversão da galactose no açúcar álcool galacitol
por uma galactose redutase dependente do NADPH
Esta redutase só existe no tecido nervoso e no cristalino
A níveis circulantes normais de galactose, a actividade do
enzima não causa efeitos lesivos.
A concentrações elevadas o galacitol cria tumefacção
osmotica do cristalino com a consequente formação de cataratas
Tratamento
O despiste desta doença permite o
tratamento precoce
A eliminação precoce da galactose
cura os sintomas
Deficiência em
galactocinase
Os principais metabolitos acvumulados são a galactose e o
galactitol
O unico sintoma é representado pelas cataratas
O tratamento é restrição da galactose
Deficiência da
UDPGalactose epimerase
Os sintomas são
semelhantes aos da deficiência em transferase
Está indicada a restrição da galactose
Frutose
Frutosuria essencial
Falta a fosfofrutocinase hepática
A situação é benigna porque não se acumulam produtos tóxicos
Parte da frutose ingerida
é fosforilada parcialmente pela
hexocinase, entrando na glicolise
Intolerância
hereditária à frutose
Causas
Falta a aldolase
Acumula-se frutose-1-fosfato em vários tecidos, inibindo a
glicogeneolise e a gliconeogenese
A frutose necessita de ATP para ser fosforilada.
A acumulação de
frutose-1-fosfato leva portanto a uma deplecção de ATP
A falta de ATP impede o fígado de realizar as suas funções
normais
principalmente por
impedir o funcionamento das bombas dependentes de ATP
Sintomas
Os sintomas só surgem após a administração de frutose ou
sacarose
A ingestão aguda de
frutose produz hipoglicemia
A ingestão crónica produz atraso de crescimento e
insuficiência hepática
Se a ingestão de frutose se mantiver surgem crises hipoglicemicas recidivantes e
insuficiências hepática e renal que podem levar à morte
Tratamento
Eliminação total da frutose, sacarose e sorbitol
Capitulo 21
METABOLISMO DOACIDO GLICURONICO
Síntese
A glicose-1-fosfato combina-se com a UTP para dar a UDPG
A UDPG oxida-se no C6 para dar o UDP-glicuronato pela
acção da UDPG deidrogenase
Esta, perdendo a UDP origina o acido glicuronico
Catabolismo
Transforma-se em L -xilulose com a formação intermediaria dos
ácidos gulonico e cetogulonico
A L-Xilulose isomeriza-se em D-xilulose que entrará no ciclo
de Dickens-Horecker

Síntese do acido ascórbico
Os primatas perderam a capacidadede sintetizar o acido
ascórbico
Os animais que o sintetizam fazem-no a partir do acido
gulonico
PATOLOGIA DOS ACIDOS URONICOS
Pentosuria idiopatica
Actividade reduzida da L-xilulose reductase
Grande excreção de pentoses pela urina, especialmente após
ingestão de acido glicuronico
É assintomática
Icterícia do
recemnascido
O acido glicuronico tem a função de se combinar com
substancias endógenas, medicamentos e tóxicos formando glicuronatos
Os glicuronatos são muito hidrosoluveis, facilitando assim a
eliminação das substancias com que se conjugaram
Esta capacidade de conjugação nem sempre existe desde o
nascimento podendo levar até 2 semanas até se efectuar
Nestes casos não há glicuroconjugação
A situação mais flagrante é a não conjugação com a
bilirrubina, surgindo icterícia, a icterícia neonatal
Sindroma de
Crigler-Najjar
Deficiência da UDP-glicuronil-transferase
Surge icterícia
Também não há conjugação com outros compostos
Capitulo 22
METABOLISMO DOS DERIVADOS DAS OSES
Desoses
Desoxiribose
Será estudada a propósito do DNA
Fucose
A síntese faz-se a partir da manose-1-fosfato
Intervêm o GTP e o NADPH
Hexosaminas
São sintetizadas a partir da fructose-6-fosfato
Ácidos sialicos
É uma síntese complexa feita a partir da glucosamina-6-fosfato
Sem comentários:
Enviar um comentário